

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Wnk, with no lysine, means that there is a lack of a conserved lysine residue in subcatalytic domain II, which is required for ATP binding [1]. Wnk kinases (WNKs) are serine/threonine protein kinases and are well conserved in many species from fungi to mammals [1]. Previous work by another group revealed that four types of WNKs are expressed in humans: Wnk1, Wnk2, Wnk3, and Wnk4 [2]. After the finding that mutations in Wnk1 and Wnk4 cause the disease pseudohypoaldosteronism type II (PHA II, OMIM no. 145260), many subsequent studies followed [3]. PHA II involves hypertension, with increased NaCl reabsorption and impaired K+ and H+ excretion [3]. Those clinical symptoms of PHA II indicate that WNKs perform roles in the kidney [4]. Consequently, early Wnk studies focused on transporters or channels located in the kidney and found that Wnk4 regulates the renal outer medullary potassium channel [5], the sodium-chloride cotransporter (NCC) [6,7], the sodium-potassium-chloride cotransporter (NKCC) [6,7,8], and claudin [9,10] which is a component of tight junctions. Thereafter, it was found that activated Wnk1 phosphorylates oxidative stress responsive kinase 1 (OSR1) and STE20/SPS1-related proline/alanine-rich kinase (SPAK) and that activated OSR1/SPAK phosphorylates the N-terminus of NKCC, which is required for activation [6,8]. WNK is activated via autophosphorylation and regulated by the presence of an autoinhibitory domain [11]. Moreover, the Wnk autoinhibitory domain is conserved in all of the WNKs [11], and the autoinhibitory domain of Wnk1 restrains not only the autophosphorylation of Wnk1 but also that of Wnk2 and Wnk4 [12].
Although the presence of autophosphorylation/autoinhibition was discovered early, the regulating mechanism of WNKs is still poorly understood [13,14,15]. Several mechanisms regulating WNKs have been revealed [13]. One is ubiquitination through scaffolding proteins such as CUL3, KLHL2, and KLHL3 [16,17,18,19,20,21,22]. Another regulating system is phosphorylation modulated by phosphatidylinositol 3-kinase/Akt (PIP3K/Akt) [23] and apoptosis signal-regulating kinase 3 (ASK3) [24,25]. The activation of WNKs in low Cl- conditions is the most direct regulation of WNKs [13]. Although the presence of SPAK/OSR1 phosphorylation induced by low Cl- was revealed early [26], it remains a mystery how WNKs sense the low Cl-, and the possibility of there being another Cl- sensor that regulates WNKs still exists. Recently, the direct binding of Cl- to a catalytic domain of Wnk1 was revealed by crystallography [27]. Nonetheless, an upstream regulator of WNKs is still not enough to explain all of the regulatory machinery.
Ca2+ is a well-known second messenger that mediates various cell functions [28]. It is well known that hypotonicity induces the elevation of intracellular Ca2+ levels through the Ca2+ channels located in the plasma membrane. In addition, Ca2+-activated Cl- channels are well defined in the salivary glands. Therefore, we hypothesized that Ca2+ might regulate the WNK-OSR1 pathway, and we found that alterations of the intracellular Ca2+ level determined the phosphorylation of OSR1 and the activity of NKCC1 in a human salivary gland (HSG) cell line.
METHODS
Materials and antibodies
HSG cell line was a gift from Dr. Kyungpyo Park in Seoul National University. Rabbit polyclonal p-OSR1 antibody was purchased from Millipore (Cat#07-2273). Rabbit polyclonal OSR1 antibody was purchased from Cell signaling (#3729). Cyclopiazonic acid was from Enzo life science Farmingdale, NY, USA). 2-Aminoethoxydiphenyl borate was from Tocris (Bristol, BS11 0QL, UK). Trypsin/EDTA, Fetal Bovine Serum, and 100X Antibiotic-Antimycotic was from Gibco (Carlsbad, CA, USA). BCECF-AM, BAPTA-AM, and Pluronic acid F-127 was from Life technologies (Carlsbad, CA, USA). Fura2-AM was from Teflabs (Austin, TX, USA). NaCl, NH4Cl was from Duksan (Ansan, Gyeonggi-do, Korea). KCl, EGTA, Glucose, and HEPES was from Amresco (Solon, OH, USA). Collagenase P was from Roche (Indiana polis, IN, USA). All other chemicals include MgCl2, CaCl2, Trypsin inhibitor, sodium pyruvate, gadolinium, lanthanum, and ruthenium red was from Sigma-Aldrich (St. Louis, MO, USA).
Isolation of salivary gland cell
ICR mice were purchased from (KOATEC, Korea). All experiments were performed on adult male ICR mice (6-8 weeks of age) that were maintained on a 12h day/night cycle with normal mouse chow and water provided ad libitum. The animal studies were performed after receiving approval of the Institutional Animal Care and Use Committee (IACUC) in Yonsei University (IACUC approval no. 2014-0067). Mice were sacrificed by cervical dislocation under CO2 anesthesia. The cells were prepared from the parotid gland and sub-mandibular gland of ICR mice by limited collagenase digestion as previously described [29]. Following isolation, the acinar cells were suspended in an extracellular physiologic salt solution (PSS), the composition of which was as follows (in mm): 140 NaCl, 5 KCl, 1 MgCl2, 1 CaCl2, 10 HEPES, and 10 Glucose, adjusted to pH 7.4 with 5 N NaOH and to 310 mOsm with 5 M NaCl.
Cell culture
HSG cell-line was maintained with high-glucose Dulbecco's Modified Eagle's medium with 10% FBS and 1X Antibiotic-Antimycotic (Gibco, CA, USA) in 95% CO2, 5% O2, 37℃ incubator. Subculture was done with Trypsin/EDTA when cells are confluent. Cells that used in the calcium imaging and NKCC1 activity measurement were cultured on the 24×24 mm size of cover glass.
Reverse-transcriptional PCR
Total RNA was extracted from isolated pancreatic acinar cells using Trizol reagent (Sigma-Aldrich) according to the manufacturer's instructions. RT-PCR was performed using a SuperScript III RT kit (Sigma-Aldrich, USA) and oligo-dTs (Fermentas, MA, USA). For PCR analysis of the following primers were used:
hWnk1 (forward: 5'-AATCCAGTGCTTCCCAGACA-3', reverse: 5'-TCTGTTGGCTGCTCACTCGAGATT-3'), hWnk4 (forward: 5'-AGCTGCGTAAAGCAAGGGAATTGG-3', reverse: 5'-TTTGTCCCATCCCTTCTCCCACAT-3'), hOSR1 (forward: 5'-AGGTTCCAGTGGGCGTCTTCATAA-3', reverse: 5'-AGGCCAGCAGAAATGAGTTCCTGA-3'), hSPAK (forward: 5'-ATTCAAGCCATGAGTCAGTGCAGC-3', reverse: 5'-GCGCTGCTCCTGTTGCTAATTCAA-3'), hNKCC1 (forward: 5'-AAGCAGTCCTTGTTCCTATGGCCT-3', reverse: 5'-GCAATGCAGCCCACCAGTTAATGA-3'), hGAPDH (forward: 5'-CGGAGTCAACGGATTTGGTCGTAT-3', reverse: 5'-AGCCTTCTCCATGGTGGTGAAGAC-3')
PCR reaction was performed using EmeraldAmp GT PCR Master Mix (Takara, Shiga, Japan). PCR condition was initiated by a 5 min incubation of the samples at 94℃, preceded by 30 (Wnk1, Wnk4), 35 (OSR1, SPAK, NKCC1, GAPDH) cycles of 30 sec at 94℃, 30 sec for annealing (Wnk1 57℃, Wnk4 60℃, OSR1 58℃, SPAK 59℃, NKCC 58℃, GAPDH 58℃), and 30 sec at 72℃. After 30 (Wnk1, Wnk4), 35 (OSR1, SPAK, NKCC1, GAPDH) cycles, samples were incubated for 10 min at 72℃ for complete extension. No reverse transcription control was performed using the same protocol, except for using reverse transcriptase.
Measurement of intracellular Ca2+
Cultured on 24×24 mm coverglass, with density of 1.5×106 cells/ml with 48 h were loaded with 4 µM Fura-2/AM and 0.05% Pluronic acid F-127 for 30 min in PSS at room temperature. Fura-2/AM fluorescence was measured at an excitation wavelengths of 340/380 nm, and emission was measured at 510 nm (ratio=F340/F380) using an imaging system (Molecular Devices, CA, USA). The emitted fluorescence was monitored using a CCD camera (CoolSNAP HQ, AZ, USA) attached to an inverted microscope. Fluorescence images were obtained at 2 sec intervals. All data were analyzed using the MetaFluor software (Molecular Devices, Downingtown, PA, USA).
Immunoblotting
Protein extracts were prepared from HSG cell-line and ICR salivary glands as follows. Cells were lysed in a buffer containing (in mm): 150 NaCl, 10 Tris (pH 7.8 with HCl), 1 EDTA, 1% NP-40, 0.1% SDS, and a protease inhibitor mixture (2 Na3VO4, 10 NaF, 10 µg/ml leupeptin, and 10 µg/ml phenylmethylsulfonyl fluoride). The samples were probed overnight with 1:2,000 dilutions of antibodies against p-OSR1, OSR1 at 4℃ and then separated by SDS-PAGE.
Measurement of intracellular pH (pHi) and NKCC activity
pHi was measured using pH sensitive fluorescent dye, BCECF-AM. Cells were loaded with 2.5 µM BCECF-AM in PSS for 30 min at room temperature. The fluorescence at excitation wavelength of 490 nm and 440 nm was recorded using a CCD camera (CoolSNAP HQ, AZ, USA). Fluorescence images were obtained at 2 sec intervals. All data were analyzed using the MetaFluor software (Molecular Devices, Downingtown, PA, USA).
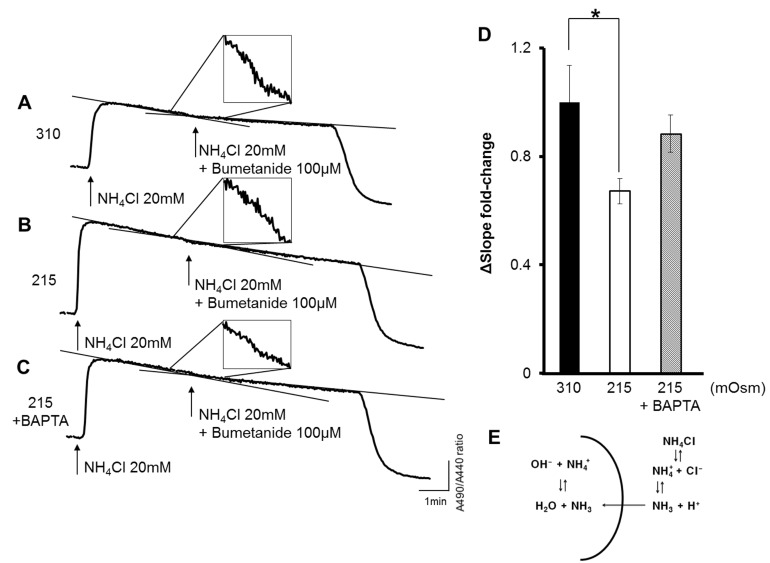
Na+-K+-2Cl- cotransporter (NKCC) activity was measured from the pHi decrease induced by the intracellular uptake of NH4+ using the methods of Evans and Turner [30] with modifications. Adding NH4Cl 20 mM in the extracellular solution induce robust increase of pHi due to diffusion of NH3 (Fig. 4E). Thereafter, pHi is getting decreased by influx of NH4+, because NH4+ acts as a surrogate of K+ in the NKCC. To discriminate only the NKCC activity, 100 µM bumetanide applied. Slope inclination was measured from 1 min to 3 min after solution change.
RESULTS
Expression and activation of Wnk kinase in the human salivary gland cell line
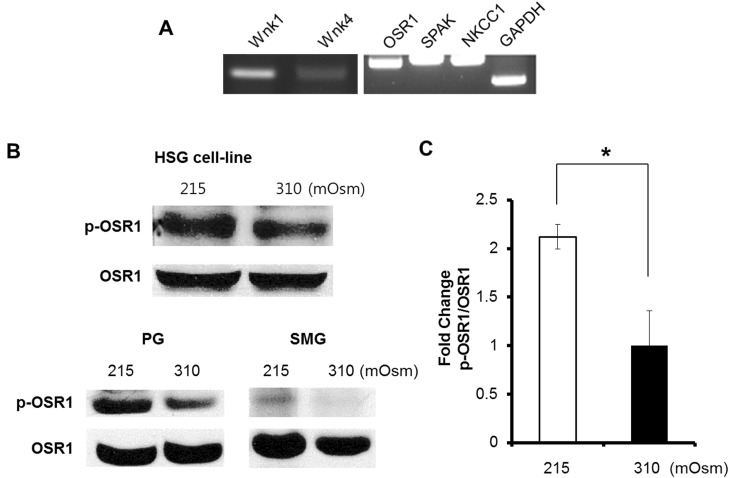
The expression of WNKs in the HSG cell line was previously unknown. Therefore, we first investigated WNK and its substrates, OSR1 and SPAK. Reverse-transcription PCR was performed, and we confirmed the mRNA expression of Wnk1, Wnk4, OSR1, SPAK, and NKCC1 in an HSG cell line (Fig. 1A). The Wnk4 expression level was lower than the Wnk1 expression level (Fig. 1A). The next step was the measurement of OSR1 phosphorylation at serine 325 by immunoblotting. The phosphorylation of OSR1 at serine 325 to form p-OSR1, the activated form of OSR1, is mediated by WNK. Hence, the elevation of p-OSR1 levels is considered indirect evidence showing the activation of WNK, because OSR1 is a main substrate of WNK. We observed an increase in the p-OSR1 level after 15 min of hypotonic (215 mOsm) stimulation at 37℃ (Fig. 1B, upper panel). The hypotonic stimulation was induced using a hypotonic solution (HS) that reduced only the NaCl content of a physiologic salt solution (PSS). We observed a p-OSR1 increase in isolated parotid gland acini and also in submandibular gland acini from an ICR mouse (Fig. 1B. lower panel). These data suggest that the HSG cell line can imitate hypotonically induced OSR1 activation.
Ca2+ signaling by hypotonic stress in the HSG cell line
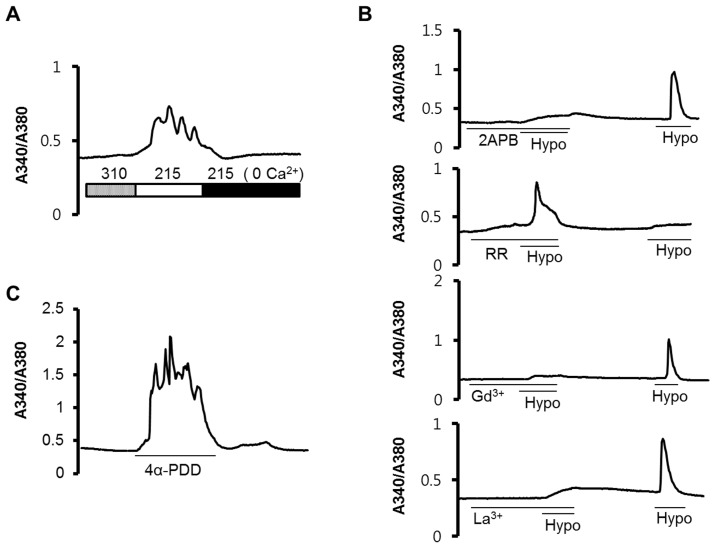
Extracellular HS treatment induced an increase in the intracellular Ca2+ concentration ([Ca2+]i), and was abolished with the depletion of Ca2+ ions in the extracellular solution (Fig. 2A), suggesting that the increased Ca2+ signal originated from the outside of the cell. To define how the Ca2+ influx was mediated by extracellular Ca2+ ions, we used 10 µM ruthenium red (RR), 100 µM 2-aminoethoxydiphenyl borate (2APB), 10 µM gadolinium (Gd3+), and 10 µM lanthanum (La3+) as blockers. After 5 min of pre-incubation with 2APB, Gd3+, and La3+, hypotonic stimulation did not evoke an elevation in the [Ca2+]i. However, RR could not inhibit the [Ca2+]i increase (Fig. 2B). When we added 10 µM 4α-phorbol 12, 13-didecanoate (4α-PDD) to the isotonic PSS (310 mOsm), we observed similar patterns of [Ca2+]i increase (Fig. 2C).
Ca2+ signaling is related to OSR1 phosphorylation
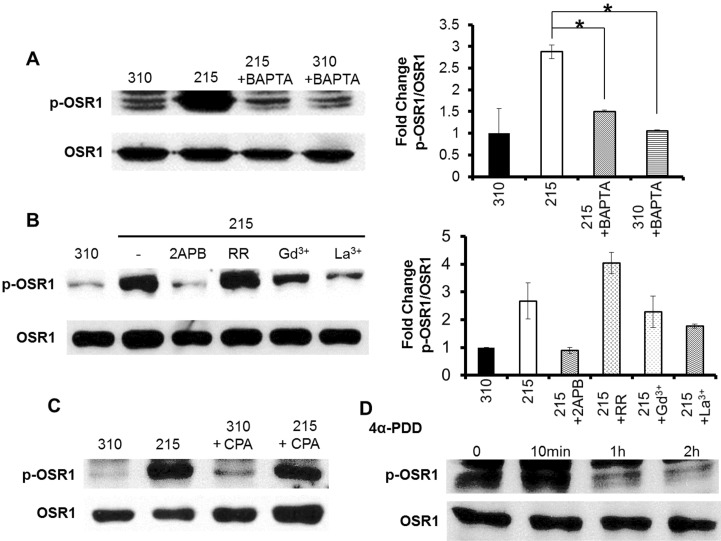
BAPTA is a high-affinity Ca2+ chelator that acts rapidly. When we loaded 25 µM BAPTA-AM, which is an acetomethoxy ester conjugate of BAPTA, 20 min prior to the hypotonic stimulation, the p-OSR1 level was reduced compared with that in a control after 10 min of hypo-osmotic stress (Fig. 3A, left panel). The fold change of the p-OSR1/total OSR1 intensity was 2.88±0.15 in 215 mOsm, 1.51±0.02 in 215 mOsm+BAPTA, and 1.06±0.01 in 310 mOsm+BAPTA (Fig. 3A, right panel). This result indicates that p-OSR1 may be regulated by upstream Ca2+ ions. The 4α-PDD-induced p-OSR1 level was also increased 10 min after treatment and diminished less than that in the control after 1 h (Fig. 3C). In agreement with the Ca2+ signaling pattern, the p-OSR1 level was reduced following a 20 min pre-incubation with the blockers 2APB, Gd3+, and La3+ (Fig. 3B). RR could not inhibit the hypotonic-induced [Ca2+]i increase in the HSG cell line (Fig. 2B, second panel from the top), and the p-OSR1 level was increased in the RR pre-incubation only (Fig. 3B). The Ca2+ release from the endoplasmic reticulum (ER) induced by 25 µM cyclopiazonic acid (CPA) could also elevate the p-OSR1 level (Fig. 3D).
NKCC1 activity is reduced under hypotonic stress
The activation of OSR1 could affect the various transporters in the plasma membrane, such as the NCC, the K+-Cl- cotransporter (KCC), and the NKCC. In the present study, we measured NKCC activity using pH changes in the HSG cells. The traces in Fig. 4A-C show the BCECF ratio of emitting wave intensity in the excitation wave-lengths 490 nm and 440 nm. Thus, the value of the BCECF ratio indicates the intracellular pH (pHi). Fig. 4E shows the scheme of the NH4Cl treatment. When 20 mm NH4Cl was added to the extracellular solution, with the substitution of NaCl to maintain the osmolarity, the influx of NH3 cause pH increase. After that, through NKCC1 activity, which carries NH4+ as a surrogate of K+, the pH in the solution acidified. Bumetanide (100 µM) can inhibit NKCC activity. Therefore, the difference in the slope inclination between the NH4Cl treatment and the bumetanide treatment was assessed to measure the NKCC1 activity by quantifying the acidification rate. The greatest slope change was with 100 µM bumetanide in the isotonic solution (310 mOsm; Fig. 4A). After pre-incubation in HS (215 mOsm) for 5 min, the fold change in the slope was 0.67±0.05 (Fig. 4B), which was statistically significant (Fig. 4D). After pre-incubation with 25 µM BAPTA-AM in HS, the decreased fold change in the slope was restored (Fig. 4C, D).
DISCUSSION
In the present study, we found that the phosphorylation of OSR1 is regulated by the [Ca2+]i. This was a unique finding of our study. In other articles, it was shown by using a kinase assay that Wnk4 kinase activity increased depending on the Ca2+ concentration in vitro, and that effect was diminished by the substitution of acidic amino acid residues such as E562 and D564 in Wnk4 [31]. Tacrolimus, which is a calcineurin inhibitor, induced Wnk3, Wnk4, and SPAK expression in mouse kidney [32]. Those previous studies support our findings that p-OSR1 is regulated by the [Ca2+]i.
The other issue in this study is that a source of increased Ca2+ triggered the phosphorylation of OSR1. When we removed the external Ca2+ from the hypotonic solution (Fig. 2A), the [Ca2+]i was reduced to the baseline. That result shows that the increased [Ca2+]i in the HSG cell line originated from the influx of external calcium. 2APB, an inhibitor of Ca2+ channels [33,34], was used to block the influx of Ca2+. 2APB also inhibits the IP3 induced Ca2+ increase. However, even though 2APB acts as an inhibitor of either IP3 induced Ca2+ increase or nonselective cation channel, the outcome of 2APB treatment is an inhibition of [Ca2+]i increase. In the results pretreatment with 2APB inhibited the elevation of the [Ca2+]i and reduced the level of p-OSR1 induced by hypotonic stress in the HSG cell line (Fig. 2B, 3B). Furthermore, nonselective cation channel blockers, like Gd3+ and La3+, also inhibited the [Ca2+]i and the p-OSR1 level under hypotonic stress in the HSG cell line (Fig. 2B, 3B). Those results indicate external Ca2+ influx mediated by Ca2+ channels sensitive to 2APB, Gd3+, and La3+ in the HSG cell line. Interestingly, treatment with 4α-PDD, which is a selective activator of TRPV4, displayed Ca2+ signaling patterns similar to those displayed by hypotonic stimulation [35,36]; TRPV4 is a well-known Ca2+ permeable channel activated by hypotonic stress [37]. Thus, a TRPV4-induced [Ca2+]i appears to be one potent channel that mediates the OSR1 pathway in the HSG cell line, but that hypothesis requires more experiments to be confirmed. Likewise, another TRP channels, such as TRPC3, and TRPM4 also involved hypotonicity stimulated Ca2+ influx in the HSG cell line, but we have not included in this paper. It was unexpected that RR could not inhibit the [Ca2+]i and the p-OSR1 level. That result suggests that RR-resistant Ca2+ channels play a role in the HSG cell line. However, to solve that remaining question about RR, another investigation is required.
The next question is whether the Ca2+ performed as an upstream regulator or as a downstream messenger of OSR1 phosphorylation. Because BAPTA is a rapid and high-affinity chelator of Ca2+, it is regarded as a highly efficient chelator that blocks the action of Ca2+ in the cell [38]. When the [Ca2+]i was chelated by BAPTA during the hypotonic stimulation of the HSG cell line, the p-OSR1 level was decreased (Fig. 3A). That suggests that Ca2+ is an upstream regulator of p-OSR1. Ca2+ is more important than the mechanical membrane stretching induced by the HS. It is inferred from the facts that the [Ca2+]i increases with 4α-PDD (Fig. 3D), and CPA (Fig. 3C) also resulted in increased p-OSR1 levels. However, the CPA induced less p-OSR1 than did the hypotonic stimulation. That could be explained by the localization of the WNKs near the plasma membrane [1].
Although, Ca2+ regulates OSR1 through phosphorylation, it is still unclear whether OSR1 is directly or indirectly regulated by the WNKs. Ca2+ was required to show the kinase activity of Wnk4. In the intact cell, however, remaining kinase activity appeared at very low Ca2+ concentrations, below 1 nM [31]. Moreover, it is well known that WNKs exhibit autoinhibition and autophosphorylation [11,12], and role of low Cl- in the activation of WNKs was revealed by crystallography [27]. Furthermore, it is well known that Ca2+-activated Cl- channels, like TMEM16A, are expressed in the apical membranes of salivary gland acinar cells [39]. Therefore, we assumed that TMEM16A activated by an elevated [Ca2+]i contributes to the local depletion of Cl- at the microdomain level, and that the depletion of Cl- leads to the activation of the WNK-OSR1 pathway. However, more investigations will be required to test that model.
The popular effectors of activated OSR1 are the NCC, the KCC, and the NKCC [13,14,15]. Furthermore, the NKCC is the essential molecule that maintains Cl- homeostasis in salivary gland acinar cells [39]. For that reason, we investigated the alteration of NKCC activity in the HSG cell line (Fig. 4). After hypotonic stimulation, NKCC activity was reduced in the HSG cell line and was recapitulated by chelating Ca2+ through BAPTA pretreatment (Fig. 4D). It is still controversial whether activated OSR1 contributes to the activation or to the inactivation of NKCC, depending on the cell type [24,40]. In terms of volume homeostasis, we think that the inhibition of NKCC requires cells to decrease the intracellular ion osmolarity to maintain the cell volume.
In conclusion, we found that Ca2+ regulates the WNK-OSR1-NKCC pathway. This is the first study to demonstrate the Ca2+-mediated WNK-OSR1-NKCC pathway in an intact HSG cell line. We hope that these results will help to clarify the regulatory mechanism of the WNK-OSR1 pathway and further our understanding of the mechanism of salivary secretion.
 XML Download
XML Download