

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In men, prostate cancer (PCa) is the second most common cancer world-wide [1]. The incidence of PCa has increased in recent years with the aging of populations. Androgens are required for the most PCas thereby PCa cell growth and proliferation is sensitive to deprivation of androgen [2]. Thus, androgen deprivation therapy (ADT) is frequently used for regression of PCas. However, the effect of ADT is temporary and the majority of patients with PCa develop resistance, leading to castration-resistant prostate cancer (CRPC). CRPCs persistently grow despite testosterone deprivation, and eventually lead to patient mortality [3]. Although it seems likely that prolonged deprivation of testosterone changes PCa context, the exact mechanism for attainment of hormone-refractory status remains ambiguous.
The androgen receptor (AR) gene encodes a 110-kDa nuclear receptor protein that regulates transcription of target genes. Androgen-dependent gene expression regulates cellular growth, differentiation, and development of male reproductive organs including prostate gland [4,5]. AR associates with genomic androgen responsive elements (AREs) harboring a 15-bp palindromic AR binding sequence (ARBS) motif [6]. In addition to this core regulatory sequence, varied composition and context of AREs are found in different cell types and genes, which thereby confer multiple regulatory mechanisms of AR-mediated gene expression [7]. Stimulation of LNCaP prostate cancer cells with androgen alters the level of ~500 transcripts screened by expression profiling [8]. However, the genes that are involved in androgen-dependent prostate cancer cell proliferation have not yet been identified.
Wnt proteins consist of 19 highly conserved glycoproteins that have been implicated in key processes including proliferation, migration, polarity determination, cell fate determination, and self-renewal of stem cells [9]. Wnt can activate different receptor-mediated signaling pathways and defects in Wnt signaling lead to developmental defects. Wnt signaling pathways can be classified as canonical, in which β-catenin is involved, or non-canonical, in which signals are β-catenin-independent. Wnt can bind to frizzled-1 (Fz-1) and low-density lipoprotein-related protein (LRP) 5 or 6 heterodimer. The binding of Wnt causes phosphorylation of LRP6 co-receptor at cytoplasmic residues by casein kinase-1 (CK-1) and glycogen synthase kinase-3 (GSK-3). Phosphorylation of LRP6 allows interaction with Axin complex, also called β-catenin destruction complex, which contains adenomatous polyposis coli (APC), CK-1, and GSK-3. Therefore, occupation of Fz-1/LRP6 with Wnt destabilizes β-catenin destruction complex, thereby enhancing the free β-catenin level to bind the T cell factor/lymphoid enhancer-binding factor (TCF/LEF) family of transcriptional factors and, promoting gene expression that is involved in growth and proliferation [10,11].
The role of Wnt/β-catenin signaling in prostate tumorigenesis has been extensively studied. Clinically, up-regulation of Wnt and β-catenin was observed in patients with prostate carcinoma [12]. In addition, forced activation of Wnt/β-catenin signaling in a mouse model promotes prostate adenocarcinoma [13]. Furthermore, genetic ablation of APC, which is an important scaffold protein for the formation of β-catenin destruction complex, leads to the development of prostatic adenocarcinoma [14]. In addition to its role in prostate cancer development, Wnt/β-catenin signaling is also important for breast tumor development [15]. Therefore, both the androgen and Wnt/β-catenin signaling pathways seem to play essential roles during prostate cancer development. However, the relationship between androgen and Wnt/β-catenin signaling has not been determined so far.
Although it has been reported that AR and β-cantenin co-localize at nucleus in castration-resistant prostate cancer patients, the role of AR in the modulation of Wnt/Fz-1 signaling pathway in hormone-sensitive prostate cancer cells is still ambiguous. In the present study, we examined androgen-dependent proliferation of PC3, VCaP, LNCaP prostate cancer cells and demonstrated that androgen-sensitive LNCaP prostate cancer cells regulates Wnt/β-catenin signaling by induction of LRP6.
Go to : 
METHODS
Reagents
Dulbecco's modified Eagle's medium (DMEM), Roswell Park Memorial Institute (RPMI), F-12K, fetal bovine serum (FBS), trypsin-EDTA, and antibiotics were purchased from Hyclone Laboratories, Inc. (Logan, UT). DAPI was purchased from Molecular Probes, Inc. (Carlsbad, CA). Testosterone and bicalutamide (an androgen receptor inhibitor) and all other reagents were high quality and were purchased from Sigma-Aldrich (St Louis, MO) unless otherwise indicated.
Cell culture
PC3, LNCaP, and VCaP cells were purchased from American Type Cell Culture (Manassas, VA) and cultured in F-12K, RPMI, and DMEM, respectively. Cells were supplemented with 10% (v/v) FBS and penicillin/streptomycin, and maintained at 37℃ in 5% CO2. To generate lentivirus, HEK293FT cells were cultured in DMEM and maintained at 10% FBS/5% CO2/37℃.
Analysis of mRNA expression
The mRNA expression was quantified by reverse transcription PCR (RT-PCR) analysis after isolation of total RNA using TRIZOL reagent, as described in the manufacturer's protocol (Life Technologies, Grand Island, NY). 1 µg of total RNA was reverse transcribed into cDNA using ImProm-II reverse transcription system (Promega Biotec.), which was then amplified by PCR using specific primers for androgen receptor (forward, 5'-ctctctcaagagtttggatggct-3'; reverse, 5'-cacttgcacagagatgatctctgc-3'), LRP6 (forward, 5'-attgacatgacaggtcgagagg-3'; reverse, 5'-ttgcatctccattgcatcggag-3') and GAPDH (forward, 5'-gagtcaacggatttggtcgt-3'; reverse, 5'-tgtggtcatgagtccttcca-3'). Equal amounts of RT-PCR products were separated on a 2% agarose gel and visualized by staining with ethidium bromide (Sigma-Aldrich, St. Louis, MO). Expression of LRP6 was quantified by real-time quantitative PCR (Q-PCR) using primers for LRP6 (forward, 5'-aggcacagagcgatcacatt-3'; reverse, catcgtctactcatttggggct-3'). Data are analyzed by 2-ΔΔCT method.
Lentiviral gene silencing
To generate lentiviral particles carrying target shRNA sequence, HEK293FT cells were co-transfected with pLKO.1 constructs (2 µg), pVSV-G (0.2 µg) and Δ8.9 (2 µg) by the calcium phosphate method as described previously [16]. Target sequences were 5'-cgcgactactacaactttcca-3' (for AR) and 5'-ggctcaaccgtgaagttatag-3' (for LRP6).
Cell proliferation assay
Prostate cancer cells (1×104) were plated on 6-well plates and proliferation in medium containing 1% FBS in the absence or presence of testosterone (10 nM) for 6 days was then assessed. Cells were fixed with 4% paraformaldehyde, and the nuclei were stained with DAPI. Cell number was counted under the fluorescence microscope at ×10 magnification (Axiovert200, Carl Zeiss, Jena).
Statistical analysis
Results are expressed as means±S.D. of three independent experiments (n=3 for each experiment). When comparing two groups, an unpaired Student's t-test was used to assess differences. p-values less than 0.05 were considered significant and indicated as * whereas p-values higher than 0.05 were considered insignificant.
Go to :
RESULTS
Testosterone is required for the proliferation of LNCaP but not PC3 and VCaP prostate cancer cells
Since androgen receptor activation plays an essential role in prostate cancer cell proliferation, we investigated the effect of testosterone on the proliferation of various prostate cancer cells. As shown in Fig. 1, PC3 cells showed the highest proliferation rate and VCaP and LNCaP cells showed similar proliferation rates in the absence of testosterone. The proliferation rates of PC3 and VCaP cells were not affected by testosterone, whereas that of LNCaP was significantly enhanced by testosterone as much as PC3 cells. These results suggest that AR plays a crucial role in LNCaP prostate cancer cells whereas AR signaling is not involved in the proliferation of PC3 and VCaP cells.
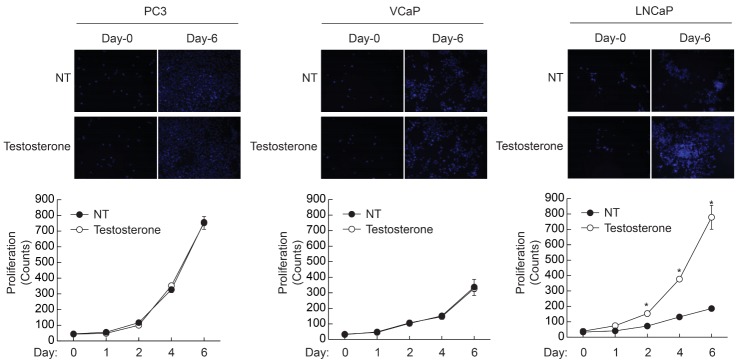 | Fig. 1Testosterone-dependent proliferation of prostate cancer cells. Prostate cancer cells (1×104/well) were plated on 6-well plates, and cultured in the presence or absence of testosterone (10 nM). Cells were stained with DAPI and images were taken at 10× magnification. Cell number was counted at the indicated time points. Data are the means±S.D. of three independent experiments (n=3 for each experiment). *Significantly different from values at the indicated time point of untreated cells (p<0.05).
|
Testosterone-dependent proliferation of LNCaP cells is abrogated by AR antagonist
Since LNCaP cells exhibited testosterone-dependent proliferation, we next examined the effect of AR antagonist on the proliferation of PC3, VCaP, and LNCaP cells. As shown in Fig. 2, stimulation of PC3 and VCaP cells with testosterone did not affect proliferation. In addition, pretreatment with an AR antagonist, bicalutamide (10 nM), had no effect on the proliferation of PC3 and VCaP cells. However, stimulation of LNCaP cells with testosterone strongly enhanced the proliferation and the increment of proliferation was abrogated by AR antagonist (bicalutamide, 10 nM). These results suggest that the AR signaling pathway is required for the proliferation of LNCaP cells; on the other hand, AR signaling is not necessary for the proliferation of PC3 and VCaP cells.
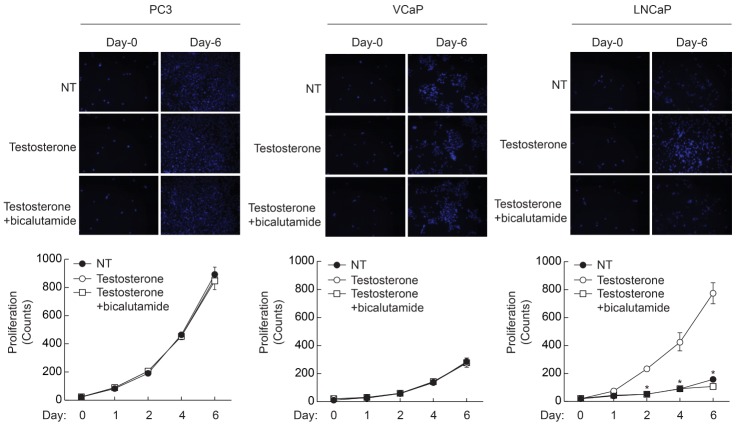 | Fig. 2Effect of AR inhibitor on the testosterone-induced proliferation of prostate cancer cells. Prostate cancer cells (1×104/well) were plated on 6-well plates, and cultured with testosterone (10 nM) in the presence or absence of AR inhibitor (bicalutamide, 10 µM). Cells were stained with DAPI and images were taken at 10× magnification. Cell number was counted at the indicated time points. Data are the means±S.D. of three independent experiments (n=3 for each experiment). *Significantly different from values at the indicated time point of testosterone treated cells (p<0.05).
|
Silencing of AR attenuated testosterone-dependent proliferation of LNCaP cells
To elucidate the role of AR in the proliferation of LNCaP cells, AR expression was silenced by infection of lentivirus containing palindromic target sequence for AR. As shown in Fig. 3, PC3 cells did not express AR, whereas both VCaP and LNCaP cells expressed the AR gene. Although VCaP cells expressed the AR gene, knockdown of AR did not affect testosterone-induced proliferation. However, knockdown of AR significantly attenuated testosterone-induced proliferation of LNCaP cells. These results suggest that activation of AR plays a crucial role in the proliferation of LNCaP cells, whereas AR is not required for the proliferation of VCaP cells even though their expression of AR is equivalent to that of LNCaP cells.
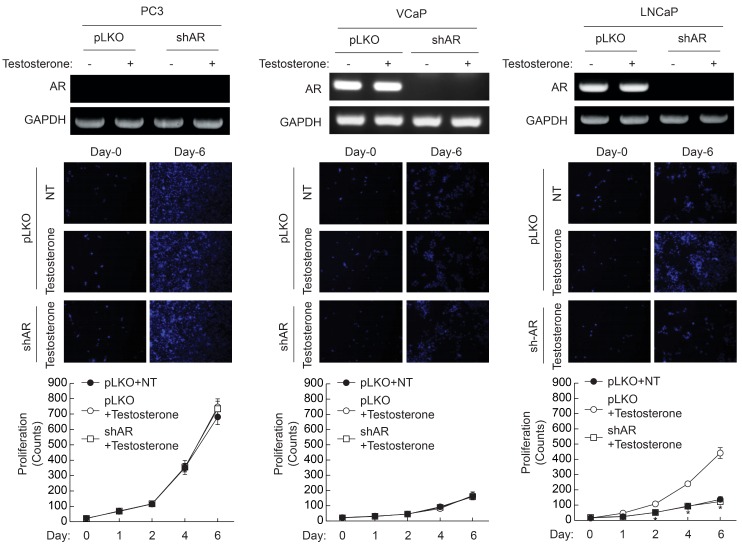 | Fig. 3Effect of AR knockdown on the testosterone-induced proliferation of prostate cancer cells. AR was silenced in each prostate cancer cell as described in "Materials and Methods". Expression of AR was verified by RTPCR (top panel). Cells (1×104/well) were plated on 6-well plates, and cultured in the presence of testosterone (10 nM). Cells were stained with DAPI and images were taken at 10× magnification (middle panel). Cell number was counted at the indicated time points (bottom panel). Data are the means±S.D. of three independent experiments (n=3 for each experiment). *Significantly different from values at the indicated time point of vector infected cells (p<0.05).
|
Expression of LRP6 is enhanced by testosterone in LNCaP cells
Since Wnt/β-catenin signaling is important for the proliferation of prostate cancer cells, we examined expression of LRP6, which is an essential co-receptor for the Fz-1 receptor to bind Wnt agonist. As shown in Fig. 4A, PC3 cells did not express LRP6. Expression of LRP6 was significantly elevated in VCaP cells; however, its expression was not altered by the stimulation with testosterone. LNCaP cells expressed less LRP6 than VCaP cells but the expression was significantly enhanced by testosterone stimulation. Testosterone-dependent expression of LRP6 in LNCaP cells was also verified by real-time Q-PCR (Fig. 4B). These results suggest that AR up-regulates LRP6 gene expression in LNCaP cells, whereas expression of LRP6 is not modulated by AR in VCaP cells.
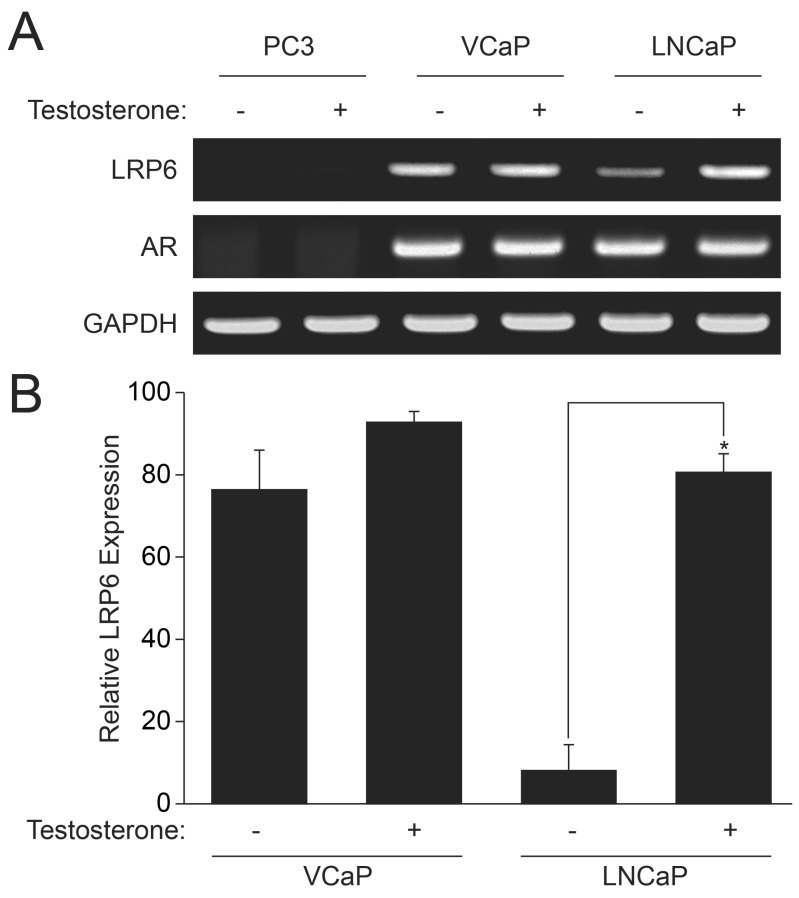 | Fig. 4Induction of LRP6 by AR stimulation. Prostate cancer cells were stimulated with testosterone (10 nM). mRNA was isolated and expression of AR, LRP6, and GAPDH was verified by RT-PCR (A). To quantify the expression of LRP-6, mRAN was quantified by real-time Q-PCR using fluorescent reporter dye (B). GAPDH was used as internal control. Data are the means±S.D. of three independent experiments (n=3 for each experiment). *Significantly different from values at the indicated time point of vector infected cells (p<0.05).
|
Silencing of LRP6 attenuates testosterone-induced proliferation of LNCaP cells
Since the expression of LRP6 was enhanced by testosterone, we examined whether knockdown of LRP6 could affect testosterone-induced proliferation. As shown in Fig. 5, infection of lentivirus containing LRP6 target sequence completely broke down LRP6 mRNA. Knockdown of LRP6 in PC3 and VCaP cells did not affect testosterone-induced proliferation, whereas stimulation of LNCaP cells with testosterone, significantly enhanced expression of LRP6. In addition, knockdown of LRP6 in LNCaP cells significantly blunted testosterone-induced proliferation. These results suggest that induction of LRP6 is required for the testosterone-induced proliferation of LNCaP cells.
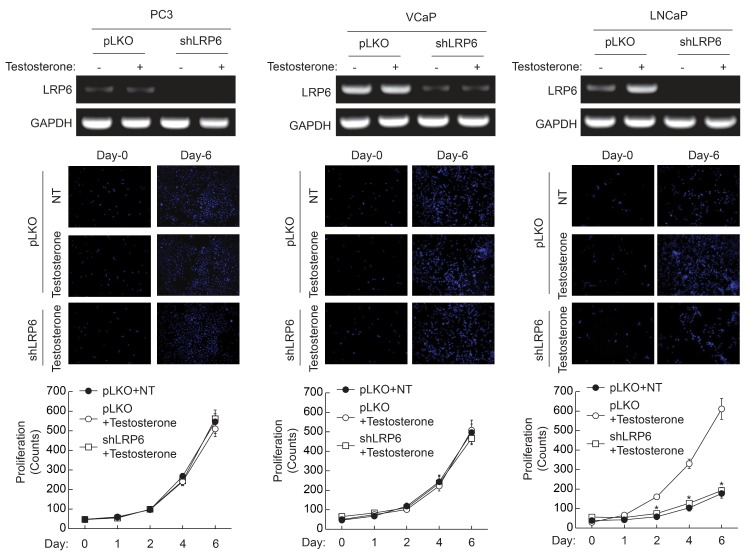 | Fig. 5Effect of LRP6 knockdown on the testosterone-induced proliferation of prostate cancer cells. LRP6 was silenced in each prostate cancer cell as described in "Materials and Methods". Expression of LRP6 was verified by RT-PCR (top panel). Cells (1×104/well) were plated on 6-well plates, and cultured in the presence of testosterone (10 nM). Cells were stained with DAPI and images were taken at 10× magnification (middle panel). Cell number was counted at the indicated time points (bottom panel). Data are the means±S.D. of three independent experiments (n=3 for each experiment). *Significantly different from values at the indicated time point of vector infected cells (p<0.05).
|
Go to :
DISCUSSION
The AR signaling pathway plays an essential role in PCa cell proliferation, survival, and differentiation. Therefore, ADT has been the first choice of advanced PCa. However, the majority of metastatic PCas progress to castrate-resistant phenotype [17]. Likewise, our results also showed that prostate cancer cells derived from metastasized tumor such as PC3 and VCaP cells showed refractory response to testosterone stimulation (Figs. 1 and 2). On the other hand, PCas isolated from supraclavicular lymph node such as LNCaP showed testosterone-dependent proliferation. It is reasonable that PC3 does not respond to testosterone stimulation since these cells do not express AR (Figs. 3 and 4). Though VCaP cells express AR gene, the effect of testosterone on proliferation was strictly blunted. Currently, several refractory mechanisms have been proposed. For example, AR gene can be amplified and subsequent overexpression of AR leads to refractory response to hormonal stimulation. It has been reported that 20% of castrate-resistant metastatic tumors exhibit AR amplification [18,19,20,21]. Moreover, overexpression of AR is reported to be sufficient to induce the castrate-resistant phenotype in preclinical models [22]. However, this mechanism seems not to be the case since expression levels of AR in both VCaP and LNCaP cells are quite similar (Figs. 3 and 4). It is noteworthy that VCaP cells show refractory response to testosterone even if the expression level of AR is equivalent to that of LNCaP cells. Another plausible refractory mechanism of VCaP cells could be mutations in the AR gene. Several mutations that allow other steroids such as corticosteroids and antiandrogen to act as agonists have been identified [23,24]. The majority of mutations are found in the ligand-binding region of the AR, altering the specificity of ligand binding by structural changes. Hence, mutant forms of AR can be activated by other hormones such as progesterone or by known AR antagonists such as bicalutamide. However, the latter mechanism is not the case here since stimulation of VCaP cells with bicalutamide did not enhance proliferation (Fig. 2).
Although ADT causes CRPC, several clinical observations imply that castrate-resistant disease still requires AR signaling [25,26]. Thus, understanding the underlying mechanism of AR-dependent prostate cancer cell proliferation is important for therapeutic application in prostate cancer cells. In the present study, hormone-sensitive LNCaP cell proliferation required testosterone as well as AR. For example, inhibition or knockdown of AR completely blocked testosterone-induced proliferation, indicating that AR signaling is important for the proliferation of LNCaP cells (Figs. 2 and 3). In addition to the AR signaling pathway, the Wnt/β-catenin signaling pathway is also known as an important signaling axis. For instance, nuclear translocation or overexpression of β-catenin is frequently detected in various prostate cancer tissues from CRPC patients [27,28]. AR directly interacts with β-catenin in the presence of dyhydrotestosterone [29]. The interaction between AR and β-catenin can be modulated by other factors in prostate cancer cells. The proximal region of the binding groove for p160 coactivators such as TIF2/glucocorticoid receptor interacting protein-1 (GRIP1) in AR interacts with β-catenin, leading to synergistic activation of AR-dependent transcription [30,31]. In addition to direct interaction of AR with β-catenin, our results showed that AR augmented Wnt/β-catenin signaling by up-regulation of LRP6 gene expression. For instance, stimulation of LNCaP cells with testosterone markedly enhanced the expression of LRP6 (Fig. 4). In addition, silencing LRP6 in LNCaP cells blunted testosterone-induced proliferation (Fig. 5). These results suggest that up-regulation of LRP6 plays a crucial role in testosterone-dependent proliferation of PCas. Thus, a positive-feed forward mechanism between AR and Wnt/β-catenin signaling pathway seems to be an important regulatory mechanism for the progression of PCa.
Go to :
 XML Download
XML Download