

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Vascular calcification can cause life-threatening organ dysfunction. Several factors, such as high phosphate levels, parathyroid hormone, vitamin D, and glucocorticoids are known to contribute to vascular calcification [1]. Previous studies have shown that vascular smooth muscle cells (VSMCs) appear to mineralize in the presence of β-glycerophosphate and undergo phenotypic differentiation into osteoblast-like cells [2,3,4]. In addition, vascular calcification is more common in patients with diabetes than in the general population [5]. Medial calcification is a common pathologic condition that occurs in diabetic patients, and may contribute to increase vascular stiffness, cardiovascular mortality, and events associated with coronary artery disease and amputations [1,6,7]. Regarding the effects of high glucose levels on the mineralization of VSMCs, high glucose stimulation in calcification medium containing β-glycerophosphate for 7 days has been reported to significantly increase calcification in bovine VSMCs in a time-dependent manner [8]. Clinically, Diabetes mellitus is characterized by chronic complications including vasculopathy under long standing hyperglycemic state [9,10]. Vascular calcification related with diabetes mellitus is also long-term process [1,5,6] and duration of diabetes is one of the important risk factors for arterial calcification [11]. However, few reports have been issued on the long-term effects of high glucose concentration with or without high phosphate levels on the progression of vascular calcification over a period of 4 weeks.
Osteoprotegerin (OPG) is a member of the tumor necrosis factor (TNF)-related family and part of the OPG/receptor activator of NF-κBligand (RANKL)/receptor activator of NF-κB (RANK) [12]. OPG is a soluble decoy receptor that competes for RANKL, and thus, prevents RANK-RANKL interactions, osteoclast differentiation, and bone resorption [13]. OPG is also able to bind and neutralize TNF-related apoptosis-inducing ligand (TRAIL) expressed by VSMCs [14,15]. Furthermore, a large number of clinical studies have reported higher OPG serum levels in association with cardiovascular outcomes in the contexts of coronary artery disease (CAD), vascular calcification, advanced atherosclerosis, diabetic complications, heart failure, abdominal aortic aneurysms, and cardiovascular mortality [16]. Regarding animal studies, one study have shown that OPG is an inducer of fibrogenesis VSMC and its actions are largely dependent on the autocrine induction of TGF-β1 [17]. Another study suggested that human full-length OPG promotes rodent VSMC proliferation and might be directly involved in pathogenetic aspects of atherosclerosis [18]. Pro-inflammatory mediators, such as, TNF-alpha and Platelet-derived growth factor (PDGF) are able to induce OPG expression in VSMCs [19,20,21]. Generally, agents that stimulate VSMC calcification also stimulate OPG production, possibly as a protective mechanism to ameliorate osteogenic or pro-apoptotic calcification processes [13,22,23]. However, it remains unclear whether OPG is a marker or rather plays a causal role in mediating or protecting against vascular injury [12]. Furthermore, the OPG/RANKL/RANK axis and TRAIL participate in vascular calcification including atherosclerosis, but their contributions have not been elucidated [24].
In this study, we evaluated the effects of high glucose and phosphate levels on the progression of vascular calcification in rat aortic smooth muscle cells and monitored changes in the expression of OPG, RANK, RANKL, and TRAIL for 2 or 4 weeks. We used BMP-7, which is known to attenuate vascular calcification [25,26,27], to detect possible changes in the expressions of OPG, RANK, RANKL, and TRAIL during the calcification of VSMCs.
METHODS
Materials
Sodium pyruvate, insulin, and fetal bovine serum (FBS) were purchased from Join Bio-Innovation (Seoul, Korea). Ascorbic acid and β-glycerophosphate were from Sigma (St. Louis, USA). Recombinant human BMP-7 (rhBMP-7) was purchased from R&D Systems (Minneapolis, MN, USA), and goat polyclonal antibodies against OPG, RANK, RANKL, TRAIL, and alkaline phosphatase (ALP), and mouse monoclonal antibody against β-actin were purchased from Santa Cruz Biotechnology (Santa Cruz, CA, USA).
Primary cell culture
Rat aortic smooth muscle cells were obtained using a modification of the explant method [28,29]. Briefly, a 4-week old Sprague-Dawley rat (male, 200~250 g) was sacrificed by forced CO2 inhalation. Permission to perform these animal experiments was obtained from the Research Institute for Convergence of Biomedical Science and Technology, Pusan National University Yangsan Hospital. The chest was rapidly opened and the descending thoracic aorta was carefully excised and incised longitudinally. Endothelium and adventitia were then removed. Small pieces of aorta (2~3 cm) were then placed in a six-well culture dish and incubated for seven days in Dulbecco's Modified Eagle's Medium (DMEM) containing 4.5 g/L glucose and 10% FBS supplemented with penicillin (100 U/mL) and streptomycin (100 µg/mL) in a humidified 95% air/5% CO2 environment at 37℃. After smooth muscle cell growth was confirmed by microscopy, aortic tissue was removed. When cells were ~70% confluent in a 100-mm dish, they were sub-cultured and maintained in DMEM (1.0 g/L glucose) containing 10% FBS supplemented with penicillin (100 U/mL) and streptomycin (100 µg/mL). The medium was replaced every two to three days. Only cells between passages 2 to 8 were used for experiments. Cells were placed in serum-depleted medium (0.1% FBS) for 24 hours to induce quiescence prior to treatment. All experiments were repeated three times using cells from different passages, and the results presented are representative of the experiments performed.
Induction of calcification
Rat aortic smooth muscle cells were allocated to a normal glucose (NG, 5.5 mmol/L D-glucose) group or a high glucose (HG, 30 mmol/L D-glucose) group. In some experiments, cells were allocated to an osmolality control (OC, 5.5 mmol/L D-glucose plus 25 mmol/L D-mannitol). Cells were cultured until confluent and then placed in calcification medium (DMEM containing 10 mmol/L sodium pyruvate, 10-7 mol/L insulin, 50 µg/mL ascorbic acid, 100 U/mL penicillin, 100 µg/mL streptomycin and 12 mmol/L β-glycerophosphate) for 2 or 4 weeks. To investigate the effect of glucose stimulation only, rat aortic smooth muscle cells were also stimulated at normal or high glucose condition without β-glycerophosphate. The medium was replaced with fresh medium every two to three days and the first day of culture in calcification medium was defined as day 0 [2]. In some experiments, to check the effects of BMP-7 on the expressions of OPG, RANK, RANKL, TRAIL, and ALP in high glucose and phosphate stimulated rat aortic smooth muscle cells, we co-treated some cells with rhBMP-7 (200 ng/mL) [27,30] for 4 weeks in high glucose condition.
Determination of calcium deposition by calcium stains
For detection of mineralization, we used two calcium stains for qualitative staining. Rat aortic smooth muscle cells were grown to subconfluence, then placed in serum-reduced medium and treated in 6 well dishes. Cells were rinsed with water, plates were drained, and 2% alizarin red solution (pH 6.0) was added. After 30-second incubation at room temperature, the plates were rinsed three times with distilled water (dH2O). Von Kossa staining was also performed as previously described [31]. In brief, rat aortic smooth muscle cells cultured in calcified medium for 2 or 4 weeks were fixed with 0.1% glutaraldehyde (Sigma) for 15 minutes at RT and washed with dH2O twice, then incubated with 5% silver nitrate (von Kossa) for 30 minutes at RT. The cells were exposed to sunlight for 2 hours until color development was complete. The sliver nitrate solution was removed and the cells were rinsed twice with dH2O. After washing, the cells were then rinsed again with dH2O. Images were obtained by scanning with an EXPRESSION 1680/Pro flatbed scanner (EPSON, Korea).
Quantification of calcium deposition
Rat aortic smooth muscle cells were cultured in six well plates, and were harvested at 2 or 4 weeks, respectively. Cells were washed twice with phosphate buffered saline (PBS) and were decalcified with 0.6 N HCl for 24 hours. The calcium content of the HCl supernatants was determined colorimetrically by the o-cresolphthaleincomplexone method (QuantiChromCacium Assay Kit, BioAssay Systems, Hayward, CA, USA) [2,4]. After decalcification, the cells were washed three times with PBS and solubilized with 0.1 N NaOH/0.1% sodium dodecyl sulfate (SDS). The protein content was measured with a Bradford Protein Assay kit (Bio-Rad, Richmond, CA, USA). The calcium content of the cell layer was normalized to protein content.
Reverse-transcription polymerase chain reaction (RT-PCR) analysis
Rat aortic smooth muscle cells (106 cells/ml) were cultured in the different conditions in a six-well plate. Total RNA was extracted with the TRIzol Reagent (Invitrogen, Carlsbad, CA, USA). cDNA was constructed from 300 ng of total RNA using MMLVRNaseH- reverse transcriptase (Superscript II; Invitrogen, Grand Island, NY, USA), and subjected to quantitative PCR (qPCR) using Taq polymerase and SYBR green (Platinum SYBRGreen qPCRSuperMix-UDG; Invitrogen) in a qPCRthermal cycler (DNA Engine Opticon; Bio-Rad, Hercules, CA, USA). Specific primers were designed as follows [32,33] : rat RANKL: 5'-TCG GGT TCC CAT AAA GTC AG-3' and 5'-CTG AAG CAA ATG TTG GCG TA-3'; rat RANK: 5'-TCG GGT TCC CAT AAA GTC AG-3' and 5'-CTG AAG CAA ATG TTG GCG TA-3'; rat OPG: 5'-ACA CAC CAA CTG CAG CTC AC-3' and 5'-TGT CCA CCA GAA CAC TCA GC-3'; β-actin: 5'-AAG TAC CCC ATT GAA CAC GG-3' and 5'-ATC ACA ATG CCA GTG GTA CG-3'; rat TRAIL: 5'-GCA CTT GAG AAA CGG AGA GC-3' and 5'-CTG GCA CTC TTC ATC AGC AG-3'; rat Bax: 5'-GCA GAG GAT GAT TGC TGA TG-3' and 5'-CTC AGC CCA TCT TCT TCC AG-3'; rat ALP: 5'-GCC CTC TCC AAG ACA TAT A-3' and 5'-CCA TGA TCA CGT CGA TAT CC-3'.
Western blot analyses
Cells were washed with cold PBS and incubated in ice-cold lysis buffer containing 5 mmol/L HEPES (pH 7.9), 150 mmol/L NaCl, 26% (vol/vol) glycerol, 1.5 mmol/L MgCl2, 1.0 mmol/L ethylendiaminetetraacetic acid (EDTA), 0.5 mmol/L dithiothreitol (DTT), and 1.0 mmol/L phenylmethylsulfonyl fluoride (PMSF), 1 µmol/L pepstatin A, 1 µmol/L leupeptin, 0.1 µmol/L aprotinin. Whole cell lysates (30 µg) were mixed with an equal volume of 4×Laemmli sample buffer. The lysates and pre-stained molecular weight markers were boiled for five minutes and separated by 10% SDS-polyacrylamide gel electrophoresis (SDS-PAGE). Proteins were electrophoretically transferred to a nitrocellulose membrane (Amersham, Little Chalfon, UK). The membrane was blocked in Tris-buffered saline containing 5% skim milk and 1% PBS-Tween-20 for one hour, and then incubated overnight at 4℃ with antibodies against OPG, RANK, RANKL, and TRAIL. The membrane was washed with PBS-Tween and then incubated with a peroxidase-labeled secondary antibody against goat IgG or mouse IgG diluted 1:3000 in 1% PBS-Tween. The membrane was washed again four times, and protein bands were visualized by enhanced chemiluminescence (LAS3000, Fuji, Japan). The band intensity was analyzed by scanning densitometry (UVITEC, Cambridge, UK).
Statistical data analysis
Data management was conducted by SPSS 18.0 software (SPSS Inc., Chicago, IL, USA). Data were expressed as mean±standard deviation (SD). The Mann-Whitney U test was used for the comparison the mean values between the two groups and Kruskal Wallis test followed by Dunn's multiple comparison was used for the comparison the mean values among three groups. Data were considered to be statistically significant at p<0.05.
RESULTS
Effects of high glucose and phosphate on the progression of vascular calcification
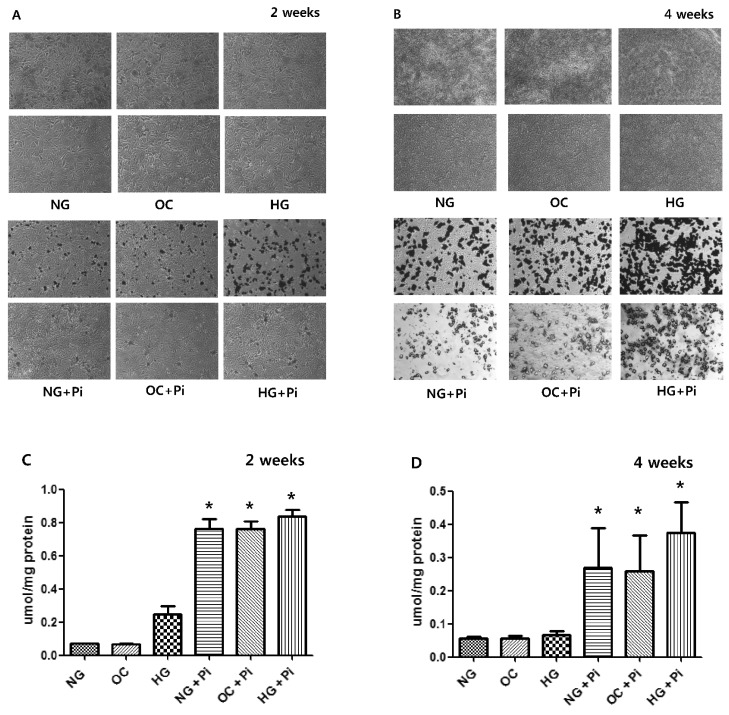
No significant change in calcium deposition was observed in RASMCs without β-glycerophosphate between high glucose group and normal glucose group after 2 or 4 weeks. However, in calcification medium with β-glycerophosphate, calcium deposition was greater in the presence of high glucose than in the presence of normal glucose after 2 weeks (Fig. 1A) and was even greater after 4 weeks (Fig. 1B). When total calcium deposition was determined by HCl extraction, no significant difference was observed between high and normal glucose group in the absence of β-glycerophosphate after 2 or 4 weeks. However, total calcium deposition was greater in high glucose group in terms of β-glycerophosphate after 2 or 4 weeks (Fig. 1C, 1D). The changes of calcium deposition in osmolality control group were similar to those of normal glucose group, suggesting that increased calcium depositions in high glucose with β-glycerophosphate were not due to the high osmolarity.
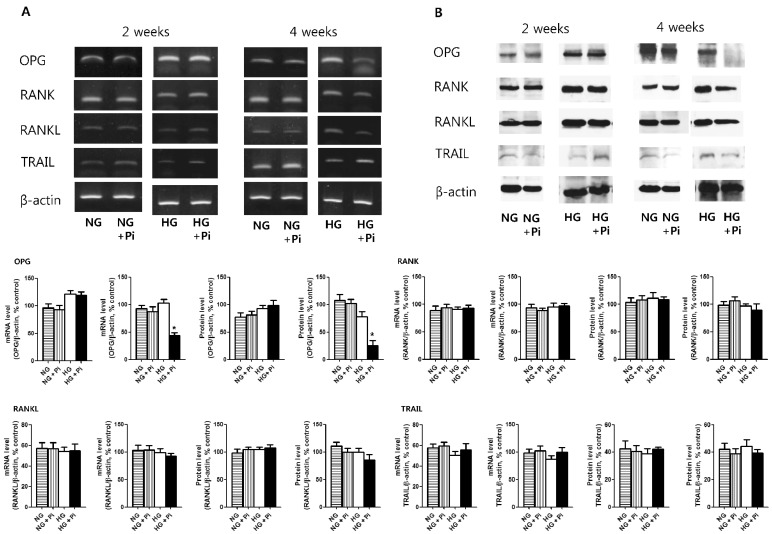
Expressions of OPG, RANK, RANKL, and TRAIL in high glucose group with β-glycerophosphate after 2 and 4 weeks
OPG mRNA and protein expressions were similar between normal glucose group and high glucose group without β-glycerophosphate after 2 and 4 weeks (Data were not shown). OPG mRNA and protein expressions were not different between high glucose group with β-glycerophosphate and high glucose group without β-glycerophosphate after 2 weeks. However, after 4 weeks, OPG mRNA and protein expressions were significantly lower in high glucose group with β-glycerophosphate than in high glucose group without β-glycerophosphate. In the case of RANK, RANKL, and TRAIL mRNA and protein expressions, no differences were observed in high glucose group with or without β-glycerophosphate for 2 or 4 weeks (Fig. 2).
Effects of BMP-7 on the progression of vascular calcification
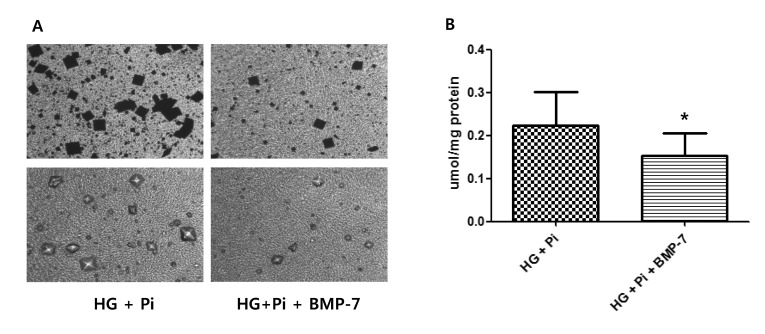
After 4 weeks of co-treatment with rhBMP-7 in high glucose group with β-glycerophosphate, calcium staining densities were attenuated and the total amount of calcium was also significantly decreased in the presence of rhBMP-7 (Fig. 3).
Effects of BMP-7 on OPG, RANK, RANKL, and TRAIL mRNA and protein expressions
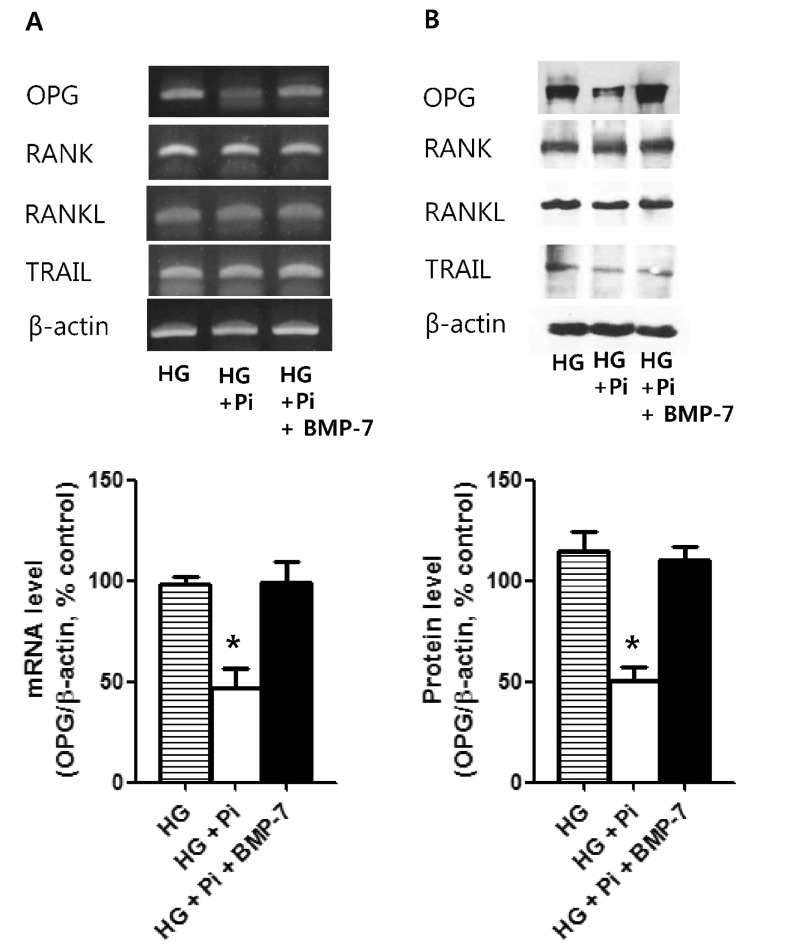
After 4 weeks of culture in high glucose group with β-glycerophosphate, mRNA and protein OPG expressions were decreased. However, when rh-BMP-7 was co-treated, these expressions were maintained. Furthermore, rhBMP-7 co-treatment had no effects on the expressions of RANK, RANKL, and TRAIL (Fig. 4).
The mRNA expressions of Bax and ALP in high glucose group with β-glycerophosphate in the presence or absence of BMP-7 after 4 weeks
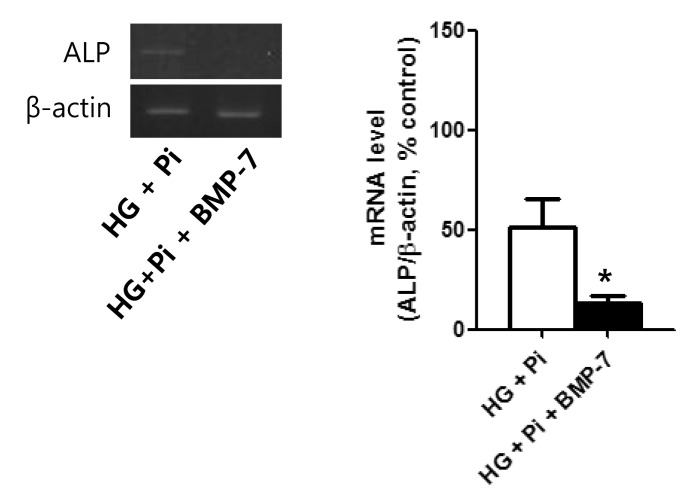
After 4 weeks of culture in high glucose group with β-glycerophosphate, ALP mRNA expression (a marker of mineralization) was decreased in the presence of rhBMP7, suggesting that the attenuating effect of BMP-7 on calcium deposition in VSMCs may be due to a reduction in osteo-induction (Fig. 5).
DISCUSSION
VSMCs are a primary target of osteogenic differentiation [3,34,35,36]. Clinically, vascular calcification related to diabetes is long-term process [1,5,6] and we aimed to investigate the long-term effect of high glucose and phosphate stimulation more than 2 weeks. The present study shows no specific increase in calcium deposition occurred in rat aortic smooth muscle cells stimulated with high glucose without β-glycerophosphate after 2 and 4 weeks. However, in calcification medium contained β-glycerophosphate, high glucose increased calcium deposition more so than normal glucose after 2 weeks and calcium deposition was more prominent after 4 weeks (as determined by von Kossa and alizarin red staining). These results suggest that chronic high glucose levels with the assistance of high phosphate accelerate the progression of vascular calcification, which is consistent with previous findings [8]. Furthermore, total calcium deposition was non-significantly greater in high glucose group with β-glycerophosphate than in high glucose group without and co-treatment with β-glycerophosphate and BMP-7 attenuated this increase in calcium deposition. We believe that more than 4 weeks is required to produce a significant increase in total calcium deposition under high glucose and phosphate conditions during the vascular calcification of VSMCs.
VSMCs are known to produce much greater amounts of OPG than endothelial cells [19] and are thought to play a significant role in the progression of vascular calcification. Furthermore, VSMCs are considered to be the major vascular producers of OPG in diseased arterial walls [37,38]. Many studies have reported that serum OPG is significantly increased in diabetic patients [39,40] and cardiovascular disease [41]. In the present study, OPG expression was observed at 2 weeks in the presence of high glucose and β-glycerophosphate, however, OPG expression was lower at 4 weeks, suggesting that OPG expression persists in the short term only. In the other hand, we found that when BMP-7, which prevent the progression of vascular calcification, was co-treated, OPG expression persisted at 4 weeks. As a possible explanation of these results, we postulate that the OPG expressions in VSMCs were increased as a defensive manner against vascular calcium deposition for short-term high glucose stimulation in calcification medium. However, for long-term stimulation (more than 2 weeks), the OPG expressions were decreasing as the vascular calcification process was more advanced with chronic high glucose stimulation. Secchiero, P et al. demonstrated that serum OPG was significantly increased in recently diagnosed diabetic patients and in steptozotocin induced diabetic apoE-null mice, but high glucose stimulations in human umbilical vein endothelial cells did not modulate OPG release when used alone or in association with TNF-α [42]. Serum OPG levels in cardiovascular disease including diabetes may be influenced by many pro-atherogenic factors besides hyperglycemia, hence, there may be some discrepancies between serum and expression levels of OPG in VSMCs. The mechanisms underlying the preventative effect of OPG on vascular calcification could be passive or cellular [12]. The anti-apoptotic effect of OPG derived by its binding of TRAIL reduces the number of apoptotic bodies that may serve as nucleation sites for passive mineralization [43]. On the other hand, vascular calcification is believed to be an active cell-mediated process resembling osteogenesis that involves the expression of bone-related proteins, such as ALP, a crucial initiator of bone mineralization [34,44,45]. In the present study, OPG mRNA and protein expressions were decreased by culture for 4 weeks in high glucose group with β-glycerophosphate, but no change in TRAIL expressions was observed, which suggests that reduced OPG activity in the long term reduces its TRAIL binding and neutralizing abilities. In addition, ALP mRNA expression, a marker of mineralization, was reduced in high glucose group with β-glycerophosphate and BMP-7 co-treatment after 4 weeks, suggesting that the effect of BMP-7 on the persistence of OPG expression in the long term period is due to reduced calcium mineralization. This finding is consistent a with previous report that a high phosphate diet and 1α, 25-dihydroxyvitamin D3 treatment in OPG-/- mice augmented severe medial calcification in VSMCs as compared with that observed in OPG+/+ controls and that apoptosis was not observed in areas of calcification. The correlation between increased aortic ALP activity and aortic calcification area in OPG-/- mice suggested an anti-calcification role of OPG via the down-regulation of ALP activity [46]. In another study conducted in OPG-/- mice, elevated serum ALP levels were found to normalize after recombinant OPG administration [23]. These results also suggest that OPG may inhibit active calcification by down regulating ALP activity.
RANK, RANKL and TRAIL showed no specific expression changes in the short or long term during calcium deposition under high glucose with β-glycerophosphate, and showed no expressions changes in high glucose group with β-glycerophosphate in the presence or absence of BMP-7 after 4 weeks of stimulation. By acting as a soluble decoy receptor for RANKL and TRAIL, OPG blocks the binding of these mediators to their cognate receptors, and thus, blocks subsequent pro-inflammatory and pro-apoptotic events [12,13,42]. Therefore, diminished OPG levels after long term stimulation with high glucose and phosphate could reduce the binding of OPG to the RANKL and TRAIL, and accelerate vascular calcification.
This study has some limitations. First, unlikely previous studies using vascular smooth muscle cells with a short-term (less than 2 weeks) stimulation, this study used primary cultured rat aortic vascular smooth muscle cells for a long-term (2~4 weeks) stimulation. Hence, there were the possibilities of being changed their shapes and properties during the stimulation period. Second, we did not demonstrate additional data on the diminished OPG levels could reduce the binding of OPG to the RANKL and TRAIL in the process of vascular calcification.
The present study showed that high glucose and phosphate induced increased vascular calcium deposition in rat aortic smooth muscle cells and OPG mRNA and protein expressions may be persisted in the short term (2 weeks). However, in the long term (≥4 weeks), OPG expressions diminished without changes in the expressions of RANKL, RANK, and TRAIL during the progression of vascular calcification. Therefore, we suggest that reductions in OPG levels might reduce the binding of OPG to RANKL and TRAIL, and thus, increase osteo-inductive VSMC differentiation. In particular, they could increase vascular mineralization, as reflected by enhanced ALP activity during the progression of vascular calcification.
 XML Download
XML Download