

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Apoptosis, a form of programmed cell death, plays an essential role in maintaining homeostasis of the immune system [1]. Deregulated apoptotic cell death leads to various autoimmune lymphoproliferative syndromes and cancer [2,3]. Apoptosis is triggered by the activation of death receptors, including Fas (Apo-1 or CD95), tumor necrosis factor (TNF), DR3 (TRAMP), DR4 (TRAIL-R1), and DR5 (TRAIL-R2) [4, 5]. Fas-induced apoptosis is a commonly encountered apoptotic pathway in various cell types. Engagement of Fas by FasL or type I-membrane protein [6,7] triggers apoptosis via the activation of specific caspases [8]. Interaction of Fas with Fas-L is required for the activation-induced cell death (AICD) of previously stimulated T cells [9-12].
TNF is a cytokine produced by various cell types, including macrophages, monocytes and lymphocytes, that induces apoptosis in response to inflammation, infection, injury, and other environmental challenges [13]. TNF has been shown to regulate cell growth, apoptosis, and differentiation, and is a mediator of inflammation as well as cellular immune responses [14-18]. Binding of TNF to its receptors, TNFR1, and less frequently to TNFR2, triggers a caspase-like signaling cascade, subsequently leading to apoptosis or nuclear factor kappa B (NF-κB) activation and cell survival [13]. Caspase-3, a cysteine-aspartic acid protease in the mitochondrial pathway of apoptosis, interacts with caspase-8 and caspase-9. Cleavage and processing of caspase-3 by the action of caspases 8, 9, and 10 leads to the activation of caspases 6 and 7 [19].
Located primarily on the outer mitochondrial membrane, the anti-apoptotic protein Bcl-2 prevents apoptosis by blocking the activation of CED-3 family proteases such as caspase-3 [20-24]. To understand the role of Bcl-2, we suppressed its expression in Jurkat T cells using short hairpin RNAs (shRNAs). Here, we report that Bcl-2 knockdown in Jurkat T cells augmented TNFR gene expression and T cell receptor (TCR)-triggered AICD but, suppressed the expression of FLIP, TNF receptor (TNFR)-associated factors 3 (TRAF3), and TRAF4. In addition, it led to increased caspase-3 cleavage and decreased NF-κB nuclrear translocation.
Go to : 
METHODS
shRNA construction and transfection
For knockdown of Bcl-2 in Jurkat T cells, four shRNAs targeting human bcl2 gene (NM_177410) were constructed using a SureSilencing shRNA plasmid (SABiosciences). The sequences of scrambled control and Bcl2-shRNAs were 5'-TGC GAC CTC TGT TTG ATT TCT-3' and 5'-GGG AGG ATT GTG GCC TTC TTT-3', respectively. Jurkat T cells were maintained in RPMI1640 containing 10% heat-inactivated fetal bovine serum (FBS), 100 U/ml penicillin, and 100 µg/ml streptomycin. For Bcl-2 knockdown, Jurkat T cells were seeded in 24-well plates at a density of 1×104 cells/well. After 24 h, cells were then transfected with Bcl2-shRNAs using FuGene 6 Transfection reagent (Roche). Efficiencies of the individual shRNAs in silencing the expression of Bcl-2 were tested, and the most effective shRNA was used in subsequent experiments. To achieve stable silencing of Bcl-2, shRNA-transfected cells were cultured in medium containing G418 for two weeks.
Determination of cell growth
Jurkat T cells (1×105 cells) were seeded in a 96-well plate and incubated for 24 h. After incubation, 0.4% Trypan Blue was added to the cell suspension, and cell numbers were estimated by counting under a light microscope. Cells stained blue were considered non-viable.
Cell viability assay
Bcl2-shRNA-transfected Jurkat T cells were incubated at a density of 5×104 cells/ml for 24 h in 96-well plates coated with 0.1 µg/ml or 1 µg/ml anti-CD3/CD28 antibodies (eBioscience). Cells were washed once with phosphate-buffered saline (PBS), resuspended in PBS containing 5 µg/ml propidium iodide (PI) (Sigma), and immediately analyzed by using a FACScalibur (BD biosciences) instrument.
Gene expression assays
Bcl-2-knockdown Jurkat T cells were incubated with plate-bound anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml) antibodies for 6 h. Total RNA was isolated using TRIzol (Invitrogen) reagent and processed for first strand complementary DNA (cDNA) synthesis. Using cDNA as a template, real-time polymerase chain reaction (PCR) was performed employing SYBR Green Master Mix (Applied Biosystems) on a LightCycler system (Roche). The relative expression levels, normalized with respect to the expression of GAPDH, were then calculated as fold induction compared to the scrambled control without any stimulation.
Western blot analysis
Bcl-2-knockdown cell lines were stimulated with plate-bound anti-CD3 antibody (1 µg/ml) and anti-CD28 antibody (1 µg/ml) for 24 h. Cells were lysed, and the protein present in the lysate was quantified using a BCA protein assay kit (Thermo Fisher Scientific). The proteins were then separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred onto a polyvinylidene fluoride (PVDF) membrane. After blocking with non-fat dried milk, the membrane was incubated with anti-Bcl-2 antibody or anti-Caspase-3 antibody (Cell Signaling) overnight, followed by horseradish peroxidase (HRP)-conjugated anti-rat IgG antibody for 2 h. Enhanced chemiluminescence (ECL) reagents were used for the detection of proteins.
Detection of nuclear NK-κB
Bcl-2-knockdown Jurkat T cells were incubated with plate-bound anti-CD3 (1 µg/ml) and anti-CD28 (1 µg/ml) antibodies for 30 min. Nuclear extracts were prepared using Nuclear Extraction Kit (Panomics) and incubated with biotinylated DNA probe (Panomics). The mixture was separated on polyacrylamide gel and the separated proteins were transferred onto a nylon membrane. After incubation with HRP-conjugated streptavidin, protein/DNA complexes were visualized using ECL reagents.
Determination of cytokine production
Scrambled- or Bcl2-shRNA-transfected Jurkat T cells were incubated with plate-bound anti-CD3 and anti-CD28 antibodies for 24 h, and the supernatants from the cultures were collected immediately. Microplates were coated overnight with purified anti-human interferon (IFN)-γ or anti-human interleukin (IL)-2 antibodies. Individual wells of the plate were blocked with a solution of 3% bovine serum albumin in PBS, and incubated overnight with the cell culture supernatants. After washing to remove unbound substances, captured cytokines were detected by applying biotinylated antibodies followed by streptavidin-alkaline phosphatase. Following washes, p-nitrophenyl phosphate (Sigma) was added to the wells and incubated for 30 min. Color developed in each well was determined by measuring optical density at 405 nm using a microplate reader (Molecular Devices).
Go to :
RESULTS
Bcl-2-shRNA suppresses Bcl-2 mRNA and protein expression in Jurkat T cells
We employed shRNAs for stable knockdown of Bcl-2 expression in human T cell lines. Four shRNA constructs targeting human bcl2 were separately transfected into Jurkat T cells, and their ability to suppress gene expression was analyzed by real-time PCR. The most effective shRNA suppressed bcl2 mRNA expression by 13.13-fold (Fig. 1A), leading to significantly lower expression of Bcl-2 protein (Fig. 1B). To examine if the silencing of bcl-2 affected cell proliferation, we cultured Jurkat T cells stably expressing Bcl-2-shRNA for 24 h and compared the cell numbers with that of control-transfected cells. In 24 h, Bcl-2-knockdown Jurkat T cells proliferated to reach 4-fold the numbers of cells initially seeded, while the numbers of control cells increased 5-fold (Fig. 1C). The ratio of non-viable cells to the total number of cells was similar for control and Bcl-2-knockdown cultures. Therefore, suppression of bcl2 expression in Jurkat T cells likely resulted in slower cell proliferation.
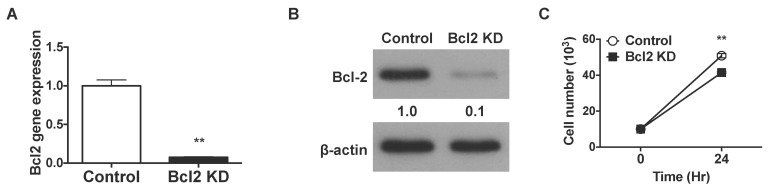 | Fig. 1shRNAs knock down Bcl-2 in Jurkat T cells. The bcl-2 gene is knocked down using shRNA in Jurkat T cells. (A) Total RNA was extracted from Bcl-2-knockdown and control Jurkat T cells and bcl-2 gene expression was analyzed by real-time PCR using GAPDH as the internal control. (B) Total protein was extracted from Bcl2-knockdown and control Jurkat T cells. Bcl-2 protein expression was analyzed by western blotting using β-actin expression as a loading control. (C) A total of 1×105 Jurkat T cells were seeded in a 96-well plate and incubated for 24 h. After incubation, 0.4% Trypan Blue was added to the cell suspension, and cell numbers were estimated by counting under a microscope. Cells stained blue were considered non-viable. Data were presented as mean±SD for triplicate determinations. Student's t test; *p<0.05; **p<0.01; and ***p<0.001 vs. control sample. All data were representative of at least three individual experiments.
|
Bcl-2 knockdown increases TCR-triggered AICD and downregulates FLIP gene expression
We stimulated AICD in Bcl-2-knockdown Jurkat T cells using anti-CD3 and anti-CD28 antibodies. Less than 10% of control T lymphocytes underwent cell death following CD3/CD28 stimulation, whereas Bcl-2 knockdown led to increased cell death (14% and 36%, respectively in the presence of 0.1 and 1.0 µg/ml antibodies) (Fig. 2A). In the absence of stimuli, both control and Bcl-2 knockdown cells showed comparable cell death rates. To investigate if Bcl-2 knockdown led to any changes in the cell death pathway, we analyzed the gene expression of Fas ligand (FasL) and Fas. Both control and Bcl-2-knockdown Jurkat T cells showed increased FasL gene expression in response to stimulation by CD3/CD28 agonists (Fig. 2B). However, the observed increase in FasL mRNA expression was lower in Bcl-2-knockdown cells than in controls. By contrast, neither Bcl-2 knockdown nor stimulation by anti-CD3/anti-CD28 antibodies altered the expression of Fas in Jurkat T cells (Fig. 2C).
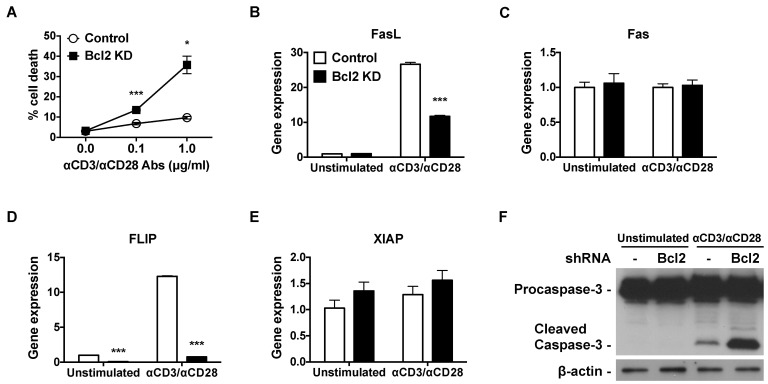 | Fig. 2Bcl-2 knockdown increases TCR-triggered AICD, downregulates FLIP gene expression, and upregulates caspase-3 cleavage. (A) Bcl-2-knockdown and control Jurkat T cells were incubated with 0.1 µg/ml or 1 µg/ml of plate bound anti-CD3 and anti-CD28 antibody for 24 h. Cells were washed with PBS, resuspended in PBS containing 5 µg/ml of PI, and analyzed by flow cytometry. (B~E) Bcl2-knockdown and control Jurkat T cells were incubated with 1 µg/ml of plate-bound anti-CD3 and anti-CD28 antibodies for 6 h. Total RNA was isolated, reverse transcribed, and gene expression was analyzed by real-time PCR. Relative gene expression levels were normalized with respect to those of GAPDH. (F) Bcl2-knockdown and control Jurkat T cells were incubated with 1 µg/ml of plate-bound anti-CD3 and anti-CD28 antibodies for 24 h. Total protein was extracted, and procaspase-3 and cleaved caspase-3 protein expression was analyzed by western blot. β-actin protein expression was used as a loading control. Data were presented as mean±SD for triplicate determinations. Student's t test; *p<0.05; **p<0.01; and ***p<0.001 vs. control sample. All data were representative of at least three individual experiments.
|
Next, we examined the gene expression of Flice-like inhibitory protein (FLIP) and X-linked inhibitor of apoptosis protein (XIAP). Stimulation of TCR/CD28 resulted in augmented expression of FLIP mRNA in control Jurkat T cells (Fig. 2D). Bcl-2 knockdown lowered the basal levels of FLIP mRNA in Jurkat T cells by 0.08-fold, and kept suppressing strongly, at a 0.76-fold-change level relative to unstimulated control cells, even under activation with anti-CD3/CD28 antibodies. Neither Bcl-2 knockdown nor TCR stimulation resulted in any significant changes in caspase inhibitor XIAP mRNA expression (Fig. 2E). We confirmed that the augmented cell death observed in CD3/CD28-activated Bcl-2-knockdown Jurkat T cells was due to increased caspase-3 cleavage (Fig. 2F). Therefore, the Bcl-2 knockdown induced a TCR-mediated AICD that was accompanied by attenuated FLIP mRNA expression.
Bcl-2 knockdown enhances the expression of TNFR and suppresses TRAF gene expression
Despite the reduced levels of FasL mRNA expression, Bcl-2-knockdown Jurkat T cells showed augmented cell death compared to control cells following TCR/CD28 stimulation. To determine the mechanisms responsible for the enhanced AICD observed in Bcl-2-knockdown cells, we analyzed the mRNA expression levels of TNFR1, TNFR2, TRAF3, and TRAF4. Following CD3 and CD28 stimulation, mRNA expression of the TNF-α receptors, TNFR1 and TNFR2, increased in Bcl-2-knockdown Jurkat T cells (Fig. 3A and B). By contrast, both basal- and agonist-stimulated expression of the inhibitors of apoptosis, TRAF3 and TRAF4, were lower in Bcl-2-knockdown cells than in control cells (Fig. 3C and D). Therefore, Bcl-2 knockdown in Jurkat T cells led to augmented expression of TNFR and suppressed TRAF3, TRAF4 gene expression.
 | Fig. 3Bcl-2 knockdown enhances the expression of TNFR and suppresses TRAF gene expression. Bcl-2-knockdown and control Jurkat T cells were incubated with 1 µg/ml of plate-bound anti-CD3 and anti-CD28 antibodies for 6 h. Total RNA was extracted and reverse transcribed, and gene expression was analyzed by real-time PCR. Relative gene expression levels of (A) TNFR1, (B) TNFR2, (C) TRAF3, (D) TRAF4 were normalized to those of GAPDH used as the internal control. Data were presented as mean±SD for triplicate determinations. Student's t test; *p<0.05; **p<0.01; and ***p<0.001 vs. control sample. All data were representative of at least three individual experiments.
|
Bcl-2 knockdown suppresses the nuclear translocation of NF-κB
The transcription factor NF-κB regulates both cell death and survival. We examined if Bcl-2 knockdown interfered with the activation of the NF-κB pathway in Jurkat T lymphocytes. Nuclear translocation of NF-κB was observed in TCR/CD28-stimulated control cells. Bcl-2 knockdown led to impaired NF-κB nuclear translocation (Fig. 4A). To confirm that Bcl-2 knockdown resulted in impaired NF-κB activation, we measured mRNA and secreted protein levels of IFN-γ as well as IL-2. Expression of IFN-γ transcripts and protein increased in agonist-stimulated control-transfected cells (Fig. 4B). Bcl-2 knockdown led to attenuated IFN-γ mRNA expression and lower IFN-γ secretion even in the presence of anti-CD3/anti-CD28 antibodies. Expression of IL-2 mRNA and protein were also found to be lower in Bcl-2-knockdown Jurkat T cells than in control-transfected cells (Fig. 4C).
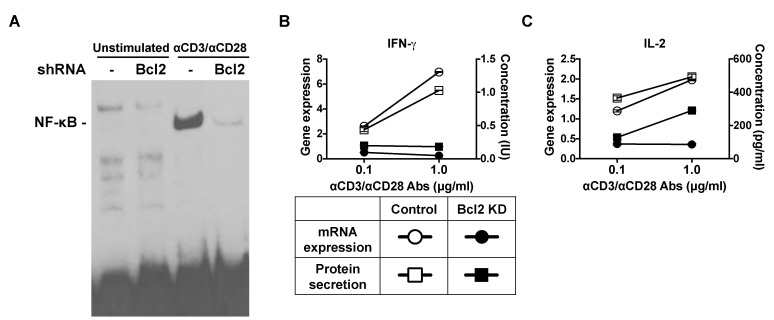 | Fig. 4Bcl-2 shRNA suppresses the nuclear translocation of NF-κB. (A) Bcl-2-knockdown and control Jurkat T cells were incubated with 1 µg/ml of plate-bound anti-CD3 and anti-CD28 antibodies for 30 min. Nuclear extracts were prepared and analyzed by using an electrophoretic mobility shift assay (EMSA). (B, C) Bcl-2 knockdown and control Jurkat T cells were incubated with 1 µg/ml of plate-bound anti-CD3 and anti-CD28 antibodies. After 24 h, supernatants were collected and analyzed for cytokines by using the enzyme-linked immunosorbent assay (ELISA) method. After 6 h, total RNA was isolated and reverse transcribed, and gene expression was analyzed by real-time PCR. Relative gene expression levels were normalized to those of GAPDH used as the internal control.
|
Go to :
DISCUSSION
Apoptosis is a developmentally and physiologically critical process that is tightly regulated by the coordinated action of diverse extracellular cues and intracellular signaling molecules [25-28]. Bcl-2 is an anti-apoptotic protein that blocks the release of cytochrome c from mitochondria. Since this molecule keeps the apoptosis pathway in check, numerous studies have focused on the roles of Bcl-2 in development, physiology, and pathophysiology. Overexpression of Bcl-2 prevents the initiation of apoptosis by blocking the efflux of cytochrome c from the mitochondria [20]. Bcl-2 overexpression in lymphoid cells suppresses apoptosis and promotes T and B cell development [29]. Furthermore, it has been shown that overexpression of Bcl-2 delays caspase-3 activation and rescues cerebellar degeneration in prion-deficient mice [30].
In our study, we knocked down Bcl-2 expression in Jurkat T cells using shRNA. Stable knockdown of Bcl-2 in Jurkat T cells led to moderate retardation of cell growth. Compared to control-transfected cells, Bcl-2 knockdown Jurkat T cells underwent significantly higher AICD following stimulation by anti-CD3 and anti-CD28 antibodies.
Engagement of Fas by FasL induces AICD in T cells. Although Bcl-2 knockdown led to enhanced agonist-induced cell death in Jurkat T cells compared to control-transfected cells, the relative expression levels of FasL mRNA followed a reversed trend. Thus, the agonist-induced increase in FasL mRNA expression was lower in Bcl-2 knockdown cells than in controls. No differences in the relative expression of Fas mRNA were observed between these cells. Therefore, we conclude that the increased apoptosis observed in Bcl-2 knockdown cells was not mediated by the Fas or FasL-related apoptosis pathways.
Caspases are crucial mediators of programmed cell death (apoptosis). Caspase-3 catalyzes the cleavage of several key proteins [31]. FLIP and XIAP inhibit programmed cell death by inhibiting caspase-3 activation. Bcl-2 knockdown in Jurkat T cells led to reduced mRNA expression of FLIP both in the absence and presence of agonists, whereas mRNA expression of XIAP remained unchanged. In addition, CD3/CD28 activation in Bcl-2-knockdown Jurkat T cells resulted in increased caspase-3 cleavage. Therefore, we conclude that knockdown of Bcl-2 induced TCR-mediated AICD, down-regulated FLIP mRNA levels, and up-regulated caspase-3 cleavage in Jurkat T cells.
TNF binding to its receptors induces apoptosis by triggering a caspase-like cascade. TNFR activation initiates signal transduction by recruiting one or more adaptor molecules known as TRAFs [32,33]. TRAF2, TRAF5, and TRAF6 induce activation of the NF-κB pathway. However, unlike other TRAFs, overexpression of TRAF3 fails to activate NF-κB [34]. Instead, TRAF3 promotes the ubiquitin-mediated degradation of NF-κB [35]. TNFR-induced apoptosis is negatively regulated by TRAF4 [36].
Compared to control-transfected cells, Bcl-2-knockdown Jurkat T cells expressed increased levels of TNFR1 and TNFR2 mRNAs, and reduced levels of TRAF3 and TRAF4 transcripts. Therefore, Bcl-2 knockdown initiated cell death by augmenting TNFR gene expression and suppressing the expression of TRAF genes.
NF-κB plays key roles in regulating both cell death and survival. We found that Bcl-2 knockdown led to altered mRNA expression of TRAF3 and TRAF4. Since it was reported that TRAFs regulate NF-κB activation [32,33,35,36], we sought to examine the possible involvement of NF-κB in the increased cell death observed in Bcl-2 knockdown cells. Compared to control-transfected cells, Bcl-2-knockdown Jurkat T cells showed impaired nuclear translocation of NF-κB following TCR/CD28-stimulation. Furthermore, Bcl-2 knockdown cells expressed reduced levels of IFN-γ and IL-2 compared to control cells under the same conditions. Taken together, Bcl-2 silencing in Jurkat T cells suppressed nuclear translocation of NF-κB and exerted an inhibitory effect on the transcription of anti-apoptotic molecules, including FLIP, TRAF3, and TRAF4 amidst unexpectedly lowering cellular FasL mRNA levels.
In conclusion, knockdown of Bcl-2 in Jurkat T cells led to elevated agonist-induced AICD, likely via a mechanism that involves enhanced TNFR1 and TNFR2 gene expression, attenuated expression of FLIP and TRAF3, increased caspase-3 cleavage, and reduced NF-κB nuclear translocation.
Go to :
 XML Download
XML Download