

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Radiation therapy is one of common conventional treatment modalities for variety of human solid tumors. Apoptosis plays an important role in cell death after DNA damage caused by radiation. In response to DNA damage caused by irradiation, p53 (53 kDa protein) activates various genes [1,2,3]. Protein products of pro-apoptotic Bcl-2 gene-family members cause cytochrome c released from the mitochondria into the cytosol and released cytochrome c activates the caspase cascade [4,5,6]. Mitochondrial apoptotic involvement could depend on signals that originate from other intracellular compartments. Namely, Ca2+ released from the endoplasmic reticulum (ER) could induce and/or play a facilitative role in the apoptotic changes [7,8,9].
Cell death has always been known to be one of the numerous cellular events triggered by an increase in intracellular Ca2+ evoked by physiological or pathological stimuli. The role of intracellular Ca2+ activity ([Ca2+]i) in apoptosis was appreciated more recently [10]. It has been reported that the expression and/or localization of Bcl-2 can modulate Ca2+ fluxes during the course of cell death [11,12,13].
Increase in intracellular Ca2+ causing apoptosis can arise from a variety of sources. Mechanisms for increasing [Ca2+]i include the entry of extracellular Ca2+ via Ca2+ channels (voltage-gated channels, receptor-mediated channels) and the transient receptor potential channel of store-operated Ca2+ influx (SOCI), or the release of stored Ca2+ form intracellular stores via IP3 receptors and ryanodine receptors in intracellular Ca2+ stores [14,15,16,17]. Once Ca2+ has served its signaling function, [Ca2+]i is lowered to resting levels to maintain intracellular Ca2+ homeostasis. Ca2+ is sequestered into intracellular stores by pumps such as the sarcoplasmic/endoplasmic Ca2+-ATPase (SERCA) or is extruded to the extracellular environment by transporters such as the Na+-Ca2+ exchanger (NCX) and the plasma membrane Ca2+-ATPase (PMCA) [14,15,17]. Mitochondria also decrease intracellular Ca2+ via the mitochondrial uniporter located in the inner mitochondrial membrane [18], although their role in regulating intracellular Ca2+ levels appears to be clearing Ca2+ in restricted microdomains such as the microenvironment of IP3 receptor channels [19].
The role of Ca2+ in promoting cell proliferation and cell death has been regarded as signaling checkpoints in cancer, which determine how these processes are remodeled in cancer [20]. Many studies have reported that a large influx of Ca2+ triggering apoptosis in cancer cells is provided by Ca2+ influx mediated by store-operated Ca2+ entry channels, which suggest a pivotal role of SOCI in apoptosis and cancer progression [21]. Moreover, the anti-apoptotic protein Bcl-2, which is commonly degraded in cancer, appears to modulate IP3-receptor Ca2+ channel activity on the ER Ca2+ stores [10,22,23]. It was also reported that the reduction in ER means that Ca2+ release is insufficient to produce apoptosis [10,24]. All these results suggest that the deregulation of cellular Ca2+ homeostasis caused under non-physiologic condition such as irradiation can elicit cell death and determine the sensitivity of cancer cells to radiotherapy. It was also suggested that ion transports may contribute to the intrinsic radio-resistance and the survival of the tumor cell, by controlling cell cycle, metabolic adaptations or DNA repair [25]. In this study, to explore the role of cellular Ca2+ metabolism in sensitivity of tumor cells to radiation, the effects of gamma (γ)-ray irradiation on cellular Ca2+ metabolism in radiosensitive RKO human colorectal cancer cells and A549 human lung cancer cells, one of known radiation-resistant cells, were examined.
METHODS
Cell culture and Irradiation of cell cultures
RKO human colorectal cancer cells and A549 human lung cancer cells were used. The cells were grown in DMEM supplemented with 10% fetal bovine serum and 1% penicillin/streptomycin. The cells were cultured in 25 cm2 plastic tissue culture flasks at 37℃ in a humidified 5% CO2/95% air atmosphere. When the cells were in exponential growth phase at a cell density of 3×106 cells/ 25 cm2 flasks, cells were irradiated with 10 Gy of γ-rays at a dose rate of 5.0 Gy/min with a 137 Cs irradiator (Cis biointernational IBL437C, France).
Measurements of [Ca2+]i
Intracellular free Ca2+ concentration was measured as described previously [26]. Cells were washed with PBS and incubated in 2 ml of buffer (0.05% trypsin and 0.02% EDTA). The cells were then resuspended in Tyrode solution (in mM: 140 NaCl, 4 KCl, 2 CaCl2, 1 MgCl2, 1 NaH2PO4, 5 HEPES, 5.5 Glucose and pH 7.4) and incubated at 37℃ with 3 µM fura-2 AM (Molecular Probe, Eugene, Oregon, USA) for 30 min and transferred to a recording chamber on an epifluorescence inverted microscope (Nikon Diaphot 300, Tokyo, Japan). Experimental solutions were superfused at a flow rate of 2 ml/min. Fluorescence intensity was measured using a cooled CCD camera (Photometrics PXL37, Tucson, Arizona, USA) and processed using the Axon Imaging Workbench v.2.2 (Axon Instrument, Foster city, CA, USA). [Ca2+]i was presented as the ratio of flurescence intensities (R340/380) excited by alternating illumination of 340 nm and 380 nm. Fluorescence intensity through 510 nm wavelength filter was collected using a cooled CCD digital camera (PXL-37, Photometrics, Tucson, AZ, USA). Experiments were done at 37℃.
Solutions
The composition of Tyrode's solution was 140 mM NaCl, 2.5 mM CaCl2, 5 mM KCl, 1 mM MgCl2, 1 mM NaH2PO4, 5 mM N-[2-hydroxyethl] piperazine-N'[2-ethanosulfonic acid] (HEPES), and 5.5 mM glucose at pH 7.4. In the 0 mM Na+/2.5 mM Ca2+ solution (Na+-free solution), NaCl was isosmotically replaced by N-methyl-D-glucamine (NMDG). 140 mM Na+/0 mM Ca2+ solution (Ca2+-free solution) was made by omitting CaCl2. To isolate NCX activity from other Ca2+ pathways, 1 µM thapsigargin (ER Ca2+-ATPase inhibitor), 5 mM caffeine (ryanodine receptor inhibitor), and 250 µM La3+ (plasma membrane Ca2+-ATPase inhibitor) were added to the superfusing solutions. The 0 Ca2+ solution, which was used to empty the internal Ca2+ stores, also contained 0.1 mM EGTA and 1 µM thapsigargin.
RESULTS
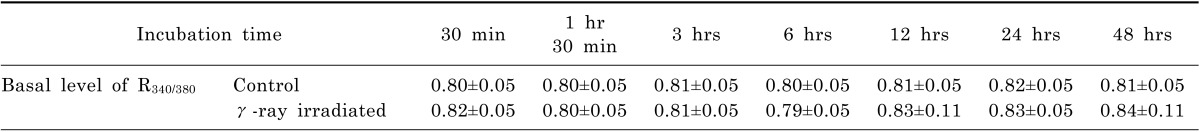
Basal level of intracellular Ca2+ activity
Basal levels of intracellular Ca2+ activities in γ-ray irradiated RKO cells were compared to those in non-irradiated control cells (Table 1). When the cells were incubated for various durations up to 48 hrs after irradiation, basal levels of R340/380 in RKO cells were not fluctuated both in the control and irradiated cells. And no difference in R340/380 was observed between the control and irradiated cells. Even when the basal levels of R340/380 were deviated most, such as in cells incubated for 48 hrs (0.81±0.05 vs, 0.84±0.11), no statistical significance was observed (p=0.14). Based on these findings, further experiments were carried out with cells which were incubated for 48 hrs after γ-ray irradiation.
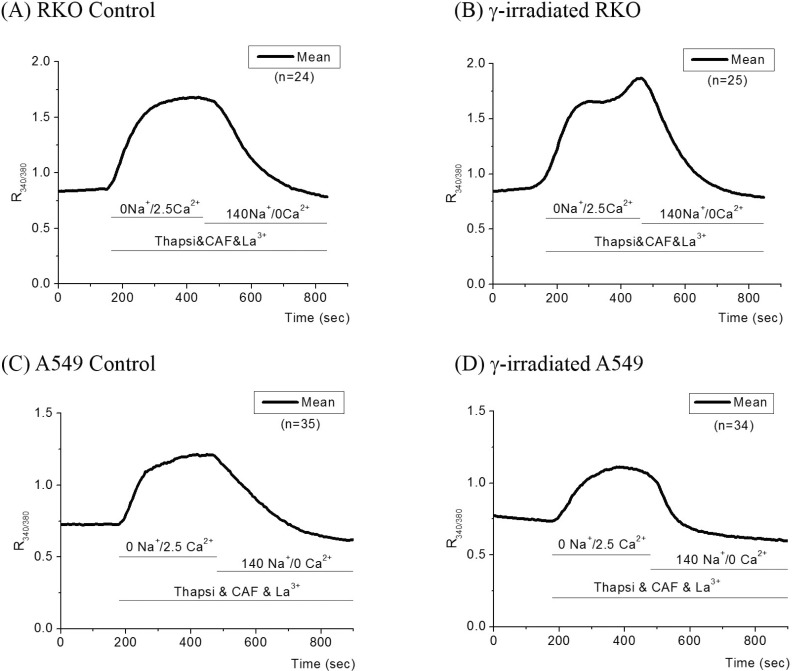
Physiological activity of NCX
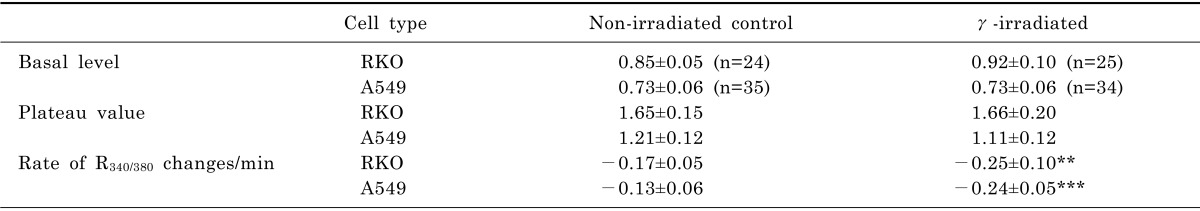
When the cells were superfused with 0 mM Na+/2.5 mM Ca2+ solution, as described in "METHODS", R340/380 in RKO cells increased to a plateau value (from 0.85±0.01 to 1.65±0.03) as shown in Fig. 1. Subsequent superfusion of 140 mM Na+/0 Ca2+ solution lowered R340/380 to the resting level with rate of R340/380 changes of -0.17±0.05 /min (Fig. 1A and Table 2). In γ-ray irradiated RKO cells, R340/380 increased to a plateau value (from 0.92±0.10 to 1.66±0.20) and an additional increase to 1.87±0.40 was followed. Subsequent superfusion of 140 mM Na+/0 mM Ca2+ solution lowered R340/380 to the resting level with rate of R340/380 changes of -0.25±0.10 /min (Fig. 1B and Table 2). The decay to the basal level of R340/380 in irradiated cells was completed faster than that of control cells (230 sec vs. 290 sec) (p<0.001).
In A549 cells, R340/380 increased to a plateau value (from 0.73±0.06 to 1.21±0.12). Subsequent superfusion of 140 mM Na+/0 mM Ca2+ solution lowered R340/380 to the resting level with rate of R340/380 changes of -0.13±0.06 /min (Fig. 1C and Table 2). In γ-ray irradiated A549 cells, R340/380 increased to plateau values (from 0.73±0.06 to 1.11±0.12). Subsequent superfusion of 140 mM Na+/0 mM Ca2+ solution lowered R340/380 to the resting level with rate of R340/380 changes of -0.24±0.05/min (Fig. 1D and Table 2). The decay to the basal level of R340/380 was also completed faster than that of control cells (p<0.0001).
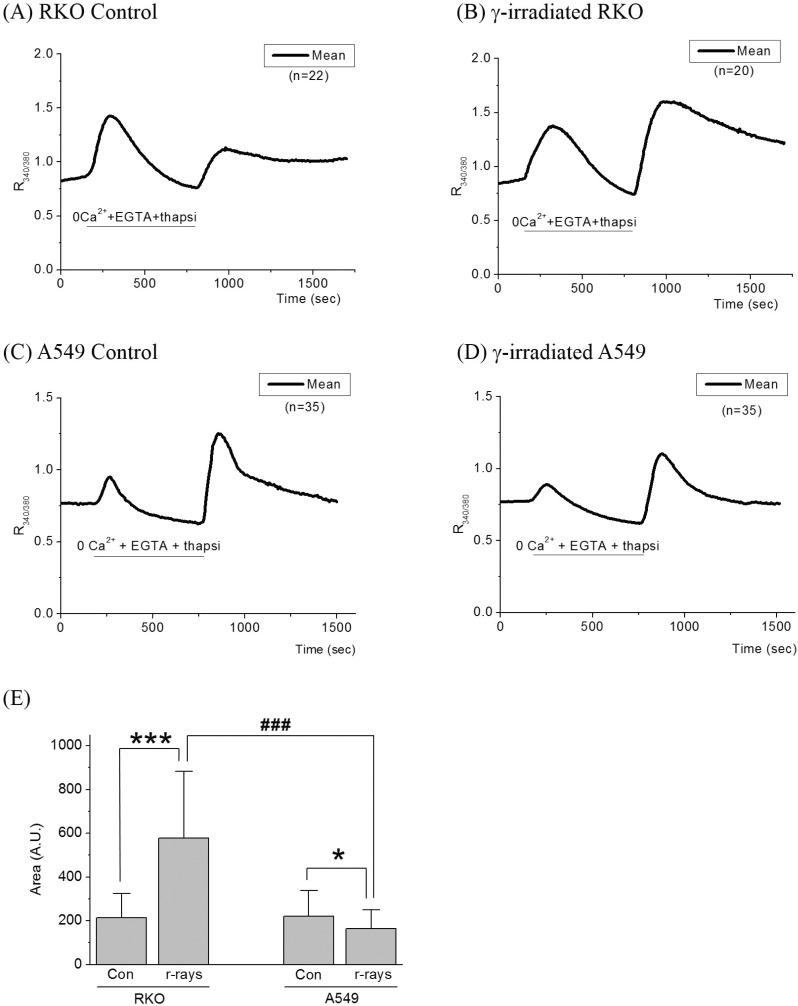
Ca2+ lnflux via SOCI
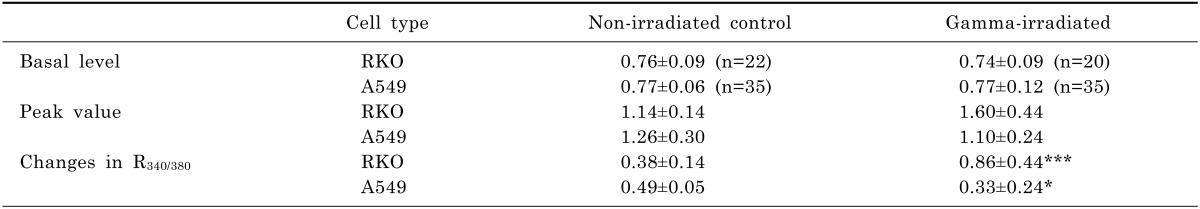
Ca2+ influx via SOCI were measured by superfusing cells with the normal Tyrode solution after empting the internal Ca2+ stores. When cells were superfused with 0 Ca2+ solution containing 0.1 mM EGTA with 1 µM thapsigargin, which empties Ca2+ out of the ER, R340/380 increased and consequently declined below the control level as shown in Fig. 2 and Table 3. In RKO cells, R340/380 increased much greater than that of A549 cells (Fig. 2A vs. 2C). In γ-ray irradiated cells, significant changes in these peaks were not observed in either cell. After the internal Ca2+ stores were emptied, superfusions of 2.5 mM Ca2+ Tyrode solution raised R340/380 from 0.76±0.09 to 1.14±0.14 in RKO cells. In γ-ray irradiated RKO cells, with superfusion of 2.5 mM Ca2+ Tyrode solution, R340/380 increased from 0.74±0.09 to 1.60±0.44 and the increments in R340/380 were much larger than those of control cells (0.86±0.44 vs. 0.38±0.14) (p<0.0001), as shown in Fig. 2B. Areas under SOCI response which approximate the amount of Ca2+ influxed by SOCI were increased by γ-ray irradiation from 213±112 to 576±304, with statistical significance (p<0.0001), as shown in Fig. 2E.
When A549 cells were superfused with 2.5 mM Ca2+ Tyrode solution, R340/380 increased from 0.77±0.06 to 1.26±0.30 (Fig. 2C). In γ-ray irradiated A549 cells, R340/380 increased from 0.77±0.12 to 1.10±0.24 as shown in Fig. 2D. The increments in R340/380 in γ-ray irradiated A549 cells were smaller than those of control cells (0.33±0.24 vs. 0.49±0.30) (p<0.05). Areas under SOCI response were decreased by γ-ray irradiation from 221±116 to 164±95, with statistical significance (p<0.05). The increment of areas under SOCI response in RKO cells by γ-ray irradiation was significantly different from that in A549 cells, which was decreased by γ-ray irradiation, as shown in Fig. 2E (p<0.0001).
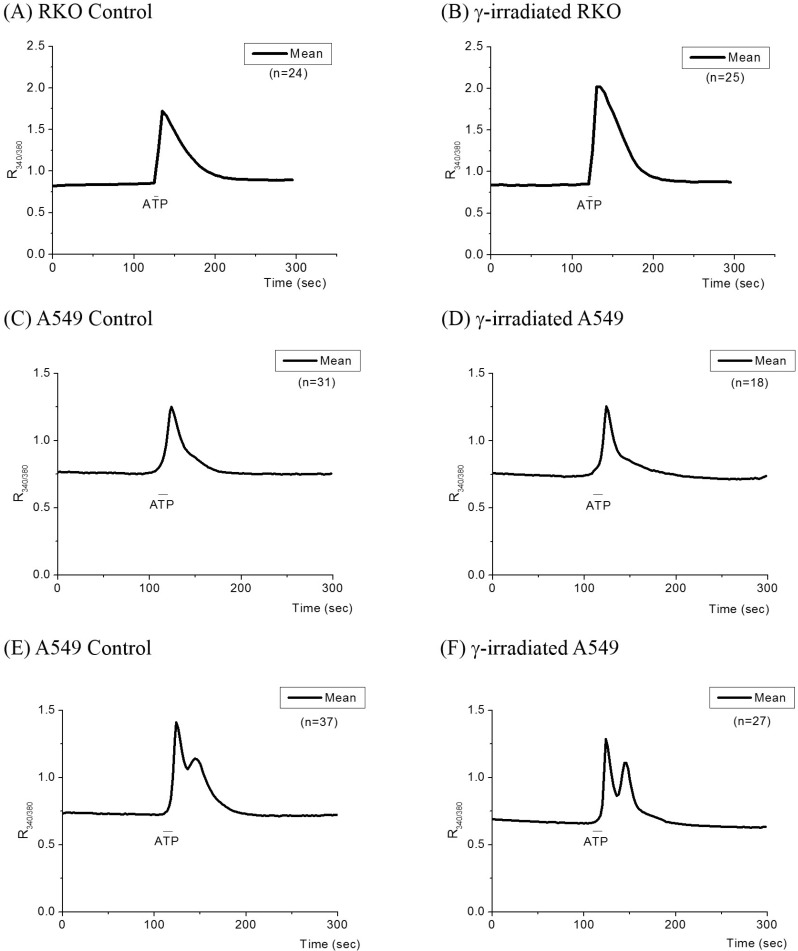
ATP-induced Ca2+ release from the ER
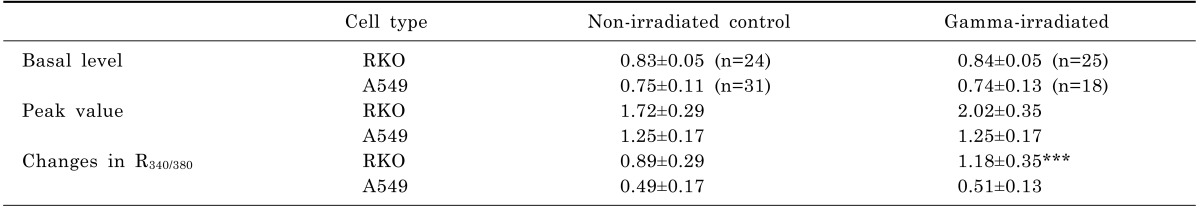
Ca2+ release from the ER was measured by applying ATP extracellularly which activates IP3 receptor channels in the ER (Fig. 3 and Table 4). When the Tyrode solution containing 100 µM ATP was applied for 10 sec, R340/380 in RKO cells increased transiently, 0.83±0.05 to 1.72±0.29, and returned slowly to the basal level (Fig. 3A). In γ-ray irradiated RKO cells, R340/380 increased from 0.84±0.05 to 2.02±0.35. The difference between the peak and basal values for R340/380 in the control cells (n=24) was 0.89±0.29 and that in the γ-ray irradiated cells (n=25) was 1.18±0.35 (p<0.0001; Table 4).
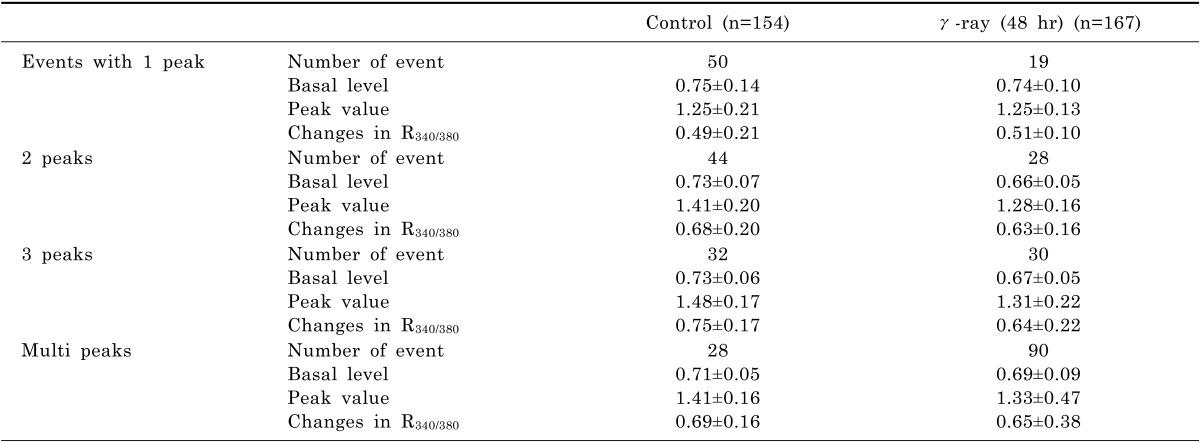
When A549 cells were superfused with the Tyrode solution containing 100 µM ATP for 10 sec, R340/380 increased transiently and returned to the basal level eliciting a single Ca2+ transient, as shown in Fig. 3C, in 50 cells out of 154 cells (32%) measured (Table 4). In other A549 cells, multiple Ca2+ transients were observed (Fig. 3E and Table 4). The amplitudes of Ca2+ transients, the differences between the peak and basal values of R340/380, were not significantly different between control cells and γ-ray irradiated cells (Table 4). However, the frequency of multiple transients was increased in γ-ray irradiated cells (Table 5).
DISCUSSION
Surge of intracellular Ca2+ causing cell death can arise from a variety of sources. [Ca2+]i can be increased by the entry of extracellular Ca2+ via SOCI or the release of stored Ca2+ form the ER via IP3 receptors and ryanodine receptors in ER membranes [14,15,16,17]. Ca2+ influx via reverse mode of NCX also contribute to [Ca2+]i increase [17,27,28].
By γ-irradiation, RKO cells begin to exit from G2/M arrest to apoptosis by 24 hrs after irradiation. Only small fractions of cells remain in G2/M phase by 48 hrs, implying that the post-mitotic apoptosis occurs by 48 hrs after irradiation [29]. During this time span, basal level of [Ca2+]i in RKO cells remained relatively unchanged (Table 1) although irradiation elicited enlargement of viable cells (data no shown). Thus experiments were done with cells incubated for 48 hrs after γ-ray irradiation.
The change in [Ca2+]i via reverse mode of NCX can be measured by blocking other cellular Ca2+ pathways as previously reported [26,27]. Irradiation does not seem to influence NCX ability to import Ca2+ into the cytosol of both RKO and A549 cells (Fig. 1). The forward mode of NCX plays a major role in clearing Ca2+ out of cytosol and can be measured by the decline of [Ca2+]i as shown in Fig. 1. Interestingly, irradiation tends to speed up the pumping activity of NCX forward mode in both cells (Table 2). It is not clear that irradiation-induced pumping activity has any physiological role in cellular metabolism. Meanwhile, to understand the cause for additional increase in [Ca2+]i via NCX over the plateau region, more information on the irradiation-induced changes in membrane fluidity is needed, since an enlargement of cells by irradiation was observed (data not shown).
Depletion or depression of Ca2+ content from ER can signal long-term cellular responses such as gene expression and programmed cell death or apoptosis [30,31] and provides a signal that activates Ca2+ entry through the SOCI channels [32,33]. Enhancement of Ca2+ entry via SOCI in RKO cells by irradiation, as shown in Fig. 2, may contribute to promotion of cell death. The enhancement of the SOCI activity may be a consequence of other cellular changes induced by irradiation, such as emptying of the ER following the increased Ca2+ release by irradiation (Fig. 3B). Irradiation may induce direct effects on SOCI-modulating proteins such as STIM and synergistic interaction of SOCI with other cellular components as reported in studies of irradiation- induced BAX interaction with SOCI [34,35].
The data of Fig. 2 provide indirect information on the ER content of Ca2+. Pre-emptying the ER to induce Ca2+ influx via SOCI can estimate the size of releasable Ca2+ pool. The results of Fig. 2 and Table 3 imply that the Ca2+ content in the ER of RKO cells is much greater than that of A549 cells. The amounts of Ca2+ released from the ER by ATP also feature the same character: RKO cells release greater amount of Ca2+ than A549 cells do (Table 4). Interestingly, γ-irradiation on A549 cells elicited decrements of ER Ca2+ content and Ca2+ influx via SOCI, while γ-irradiation on RKO cells resulted in enhancements of Ca2+ influx via SOCI (Fig. 2). These results, along with enhanced Ca2+ release from the ER by ATP in RKO cells as shown in Fig. 3, can provide possible explanation for distinct difference in cell death between RKO and A549 cells. Assuming that Ca2+ flux from the ER promotes cell death [20], enhanced Ca2+ release from the ER in RKO cells by γ-irradiation may explain radio-sensitivity of RKO cells. Unchanged Ca2+ release from the ER may be one of possible mechanisms for radiation resistivity of A549 cells. These observations are well supported by other reports stating that Ca2+ released from the reduction in ER is not sufficient to produce apoptosis [10,24].
Not surprisingly, it was found that the activity of Ca2+ transporters of A549 cells investigated in this study was not as much affected by γ-irradiation as that of radiosensitive RKO cells. However, γ-irradiation increased the incidence of multiple Ca2+ peaks in A549 cells which suggests that Ca2+-induced Ca2+ release mechanism was activated by γ-irradiation (Table 5). To clarify the involvement of this Ca2+-induced Ca2+ release mechanism in radiation-induced Ca2+ deregulation, further study with immunochemical and molecular biological methods will be needed [36]. However, the resting values of [Ca2+]i were not increased by multiple Ca2+ transients (Table 4). The results of Table 2 and 3 also support the theme that γ-irradiation does not affect intracellular Ca2+ metabolism of A549 cells and these cells may not employ the Ca2+-activated cellular process of cell death. In conclusion, these results suggest that γ-irradiation enhances the cellular Ca2+ metabolism in radiation-sensitive RKO cells and elicits programmed cell death. The results of this study may provide further understanding of the role of Ca2+ in promoting cell death and the opportunities for therapeutic intervention of cancer.
 XML Download
XML Download