

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Protease-activated receptor (PARs) are a group of G-protein coupled receptors (GPCRs) with seven transmembrane domains. To date, four members of the PAR family (PAR 1-4) have been identified [1]. The activation of PARs by proteases involves cleavage of the amino terminal sequence of the PAR extracellular N-terminal domain at specific sites. The new N-terminus exposed by cleavage acts as a 'tethered ligand' and binds to a site located in the second extracellular loop, triggering G-protein coupling and intracellular cell signaling.
Recently, it has been reported that PAR-2 is highly expressed on endothelial cells and in vascular smooth muscle, suggesting a crucial role in regulating vascular tone [2,3,4,5]. PAR-2 activation by receptor-activating peptides suppresses blood pressure in anesthetized rats or mice in vivo [6,7,8,9,10,11]. This hypotension is only partially inhibited by L-NAME, suggesting that a nitric oxide (NO)-independent mechanism and a NO-dependent mechanism are involved in the hypotension induced by PAR-2 activation [6,12]. In addition, hypotension induced by PAR-2 activation is maintained for around 2~3 min and the ganglion-blocking agent chlorisondamine significantly increases the duration of hypotension induced by PAR-2 activation, suggesting that PAR-2-induced hypotension may be maintained by modulating peripheral sympathetic tone [8,9,13]. However, a detailed mechanism for the prolonged duration of PAR-2-induced hypotension has not yet been elucidated.
It is well documented that voltage-gated N-type Ca2+ channels (ICa-N) play an important role in regulating peripheral sympathetic tone in postganglionic sympathetic neurons [14,15]. In fact, the decrease in blood pressure activates a peripheral sympathetic reflex. This enhanced peripheral sympathetic output increases Ca2+ influx through ICa-N located in peripheral sympathetic nerve terminals, that in turn triggers the release of noradrenaline (NA) from peripheral sympathetic nerve terminals. Therefore, any substance that modulates ICa-N activity can affect NA release from peripheral sympathetic nerve terminals and significantly alter sympathetic tone. It is therefore possible that PAR-2-mediated hypotension is associated, in part, with the inhibition of peripheral sympathetic output by modulating ICa-N that are located on sympathetic nerve terminals.
To investigate this hypothesis, we examined the effects of PAR-2 activation on contraction evoked by electrical field stimulation (EFS) in superior mesenteric arterial strips, which represents peripheral vascular contraction mediated by an increase in peripheral sympathetic output. We also examined the effects of PAR-2 agonists on EFS-evoked NA outflow to determine the effect of PAR-2 activation on NA release on peripheral sympathetic nerve terminals. Our results demonstrate that PAR-2 may induce hypotension by suppressing peripheral vascular sympathetic tone via the modulation of ICa-N located in peripheral sympathetic nerve terminals in rat SCG neurons.
METHODS
Animals used
Male Sprague-Dawley (SD) rats were used in all of the experiments conducted. All procedures were performed in accordance with protocols approved by the Institutional Animal Care and Use Committee of Yonsei University Health System (approval reference number: 2014-0052).
Measurement of isometric tension in rat mesenteric artery strips
Mesenteric artery strips were prepared from Male SD rats (250~350 g), as described previously [16]. Rats were deeply anesthetized with isoflurane (3%, 2~3 min) administered by inhalation in a closed chamber. To avoid unintended PAR-2 activation by coagulation factors in blood, we removed the blood from blood vessels by perfusing the rat transcardially with cold Dulbecco's phosphate buffer saline (D-PBS, composition given in the Supplementary information) for 15 min under the maintained isoflurane anesthesia. Next, the first and second branches of the mesenteric arteries were excised and helical strips of these arteries were prepared. To avoid possible influences of endothelium-derived factors, the endothelium of the strips was removed by gentle rubbing of the endothelial surface with forceps. Endothelium-denuded superior mesenteric arterial strips were used in all of the isometric contraction measurement performed. The ends of the tissue strip were tied with thin threads. One end was connected to a force-displacement transducer to monitor muscle tension and the other end was attached to a tissue holder. Strips were bathed in Krebs-Henseleit (KH) solution maintained at 37℃ and were continuously aerated with 95% O2 and 5% CO2. Muscle strips were stretched passively to impose 700 mg on the resting length and equilibrated for 60 min.
EFS
Most of the arterial segments were placed between stimulating electrodes made of platinum. Square wave pulses of 5 V with a duration 0.2 ms were delivered at a frequency of 1 Hz in 10 s trains at 5 min intervals [17,18,19]. The stimulus pulses were delivered using PowerLab (ADInstruments, Colorado Springs, CO).
NA assay
Rat mesenteric artery strips were preincubated for 30 min in 1 ml of KHS at 37℃ and continuously gassed with a 95% O2~5% CO2 mixture (stabilization period). Samples of the superfusion Krebs solution were collected before the EFS and during the EFS. Then, the tissue segments were superfused with drug for 30 min before EFS. Samples were frozen in liquid nitrogen and stored at -0℃ until the assay was performed. To measure the release of noradrenalin, we used a noradrenaline research EIA (Labor Diagnostica Nord, Gmbh & Co., KG, Nordhon, Germany).
Preparation of superior cervical ganglion neurons
Superior cervical ganglion neurons (SCG) neurons were enzymatically dissociated according to a previously described, modified method [19]. The neurons were plated on poly-l-lysine-coated 12 mm glass cover slips and incubated in a humidified incubator with 95% air, 5% CO2. Neurons were used within 12 h after plating.
Electrophysiology
Voltage-gated Ca2+ currents (ICa) were recorded using conventional whole-cell techniques. Electrode resistances varied from 3~5 MΩ when filled with internal solution. Measurements were performed using an Axopatch 200 A patch-clamp amplifier (Molecular Devices, Sunnyvale, CA). Voltage and current commands and the digitization of membrane voltages and currents were controlled using a Digidata 1322 A interfaced with Clampex 10.2 of the pClamp software package (Molecular Devices, Sunnyvale, CA), on an IBM-compatible computer. Data was analyzed using Clampfit (Molecular Devices, Sunnyvale, CA) and Prism 5.0 (GraphPad, San Diego, CA). The cells were moved from the incubator to a recording chamber, visualized using an inverted microscope, and subjected to voltage clamp experiments, using the whole cell technique. Currents were low-pass filtered at 2 kHz using the four-pole Bessel filter of the amplifier. Capacitance (Cm) values were automatically calculated during recordings by the pClamp 10.2 software. Multiple independently controlled syringes served as reservoirs for a gravity-driven fast drug perfusion system. Switching between solutions was accomplished by manually controlled valves. All experiments were conducted at room temperature.
Solutions and drugs
The Krebs-Henseleit solution (KH solution) of the following composition as modified by Asano et al (in mM) [20] : 119 NaCl, 4.6 KCl, 2.5 CaCl2, 1.2 KH2PO4 1.2, 1.5 MgSO4, 25 NaHCO3, 11 glucose. The KH solution was continuously aerated with 95% O2 and 5% CO2. The internal (pipette) solution contained the following (in mM): 140 CsCl2, 1.2 MgCl2, 4 MgATP, 0.4 Na2GTP, 10 phosphocreatine, 10 HEPES, and 10 EGTA; the solution was adjusted to pH 7.2 with CsOH. The external (bath) solution contained (in mM) 155 tetraethylammonium (TEA)-Cl, 2.5 CaCl2, 1.2 MgCl2, 14 glucose, and 10.5 HEPES; the solution was adjusted to pH 7.4 with TEA-OH. ω-conotoxin GVIA (CgTx) was purchased from Alomone (Alomone Laboratories, Jerusalem, Israel). SLIGRL-NH2 (SL-NH2, a synthetic PAR-2 agonist peptide) and LRGILS-NH2 (control peptide with s reverse sequence) [21] were purchased from COSMO Genetech (COSMO Genetech Inc., Seoul, Korea). All other drugs were purchased from Sigma-Aldrich Chemicals. All drugs were dissolved in distilled water as stock solutions (1~100 mM).
Data analysis
Data are presented as the means±SEM, with the number of experiments given within parentheses. The concentration-response curves of trypsin and SR-NH2 for ICa inhibition were calculated by fitting to a single-site binding isotherm with least-squares nonlinear regression using Prism 5.0 (GraphPad, San Diego, CA). We used unpaired Student's t-tests to compare data from two groups. Differences were considered statistically significant at p<0.05.
RESULTS
Characteristics of EFS-evoked contractions in endothelium-denuded rat superior mesenteric arterial strips
The contractile ability of the preparation was tested by perfusing a segment of the arterial strips with a 70 mM high-K+ solution (equimolar substitution of Na+ with K+) (data not shown). To avoid the possible influences of endothelium-derived factors, the endothelium of the strip was removed by gentle rubbing of the endothelial surface with a forceps. The efficiency of this method was measured by the lack of relaxation elicited by acetylcholine in NA-contracted arteries (Supplmental Fig. 1).
As shown in Supplmental Fig. 1, EFS evoked contraction in superior mesenteric arterial strips. The contraction evoked by EFS was completely blocked by 1µM TTX, a potent Na+ channel blocker, and 1µM prazosin, an α1-selective adrenergic antagonist [22,23]. This indicates that the EFS-evoked contractions of the superior mesenteric arterial strips is neurogenic, and mediated via the release of NA from sympathetic nerve endings, leading to the activation of α1-adrenoceptors in vascular smooth muscle (Supplmental Fig. 1A and B) [24,25]. In addition, 1µM ω-CgTx, a selective ICa-N blocker [24], suppressed significantly EFS-evoked contraction by 65.7±1.8%, suggesting a predominant role for ICa-N in regulation of peripheral sympathetic vascular tone (Supplmental Fig. 1C).
PAR-2 agonists suppress EFS-evoked contraction in endothelium-denuded rat superior mesenteric arterial strips
We examined the effects of PAR-2 agonists on the contractions of rat superior mesenteric arterial strips elicited by EFS. As shown in Fig. 1A and C, trypsin (30 nM) reduced the amplitude of EFS-evoked contractions by 44.1±5.4%. However, 30 nM boiled trypsin (BT) didn't have any effect on the contraction evoked by EFS in superior mesenteric arterial strips (Fig. 1A and C). Likewise, 100 µM SL-NH2, a potent PAR-2 activating peptide, decreased contraction amplitude by 41.9±6.8% (Fig. 1B and D). However, 100µM LR-NH2, a control reverse peptide, had little influence on the amplitude of EFS-evoked contractions in superior mesenteric arterial strips (Fig. 1B and D). The IC50 of trypsin on EFS-evoked contractions was 8.6±3.3 nM (Fig. 1E). Additionly, the IC50 of SL-NH2 was 32.2±4.5µM (Fig. 1E). The concentrations at which trypsin and SL-NH2 inhibited EFS-evoked contractions sub-maximally were about 30 nM and 100µM, respectively. These concentrations, therefore, were used in all subsequent experiments.
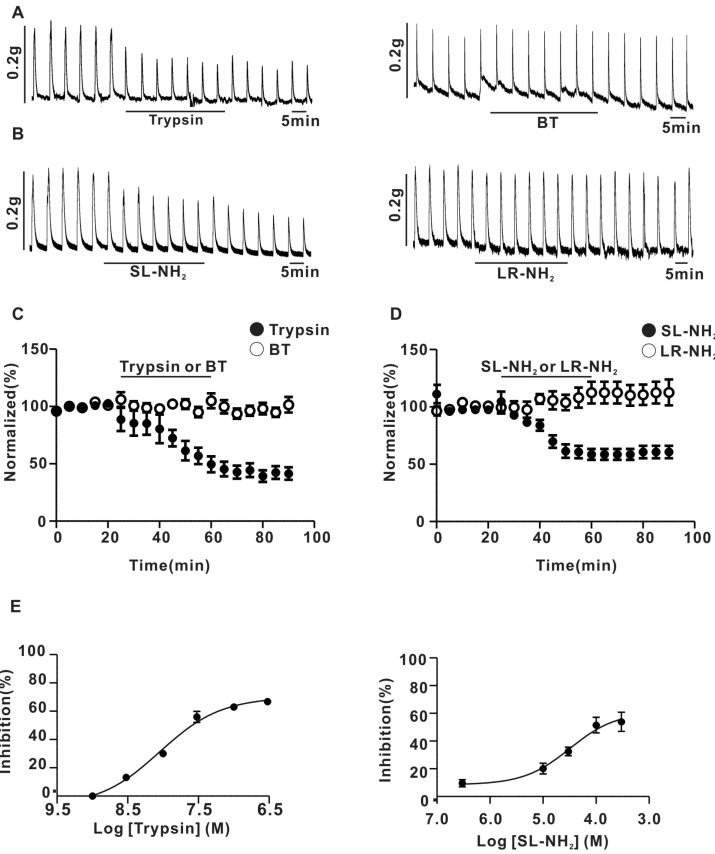
Fig. 1
Effect of PAR-2 agonists on EFS-induced contraction in endothelium-denuded rat superior mesenteric arterial strips. (A) a representative trace of inhibition of EFS-induced contraction by application of 30 nM trypsin (left) or 30 nM BT (right) (B) a representative trace of inhibition of EFS-induced contraction by application of 100µM SL-NH2 (left) or LR-NH2 (right) (C) summarized time course of trypsin (0.17±0.04 g for control, 0.10±0.03 g for trypsin, n=6, p<0.05) or BT (0.9±3.5% inhibition; 0.10±0.02 g for control, 0.10±0.03 g for BT, n=6, p>0.05) effects on EFS-induced contraction. (D) summarized time course of SL-NH2 (0.11±0.04 g for control; 0.07±0.01 g for SL-NH2, n=6, p<0.05) or LR-NH2 (2.6±3.1% inhibition; 0.6±0.1 g for control; 0.6±0.1 g for LR-NH2, n=6, p>0.05) effects on EFS-induced contraction. (E) right, Concentration-response curves for trypsin. Left Concentration-response curves for SL-NH2.
![]()
To exclude the possibility that the inhibition of EFS-contraction by PAR-2 agonists was mediated by direct inhibition of vascular smooth muscle contraction, we investigated the effects of PAR-2 agonists on contraction evoked by externally applied NA in endothelium-denuded superior mesenteric arterial strips. As shown in Fig. 2, externally applied NA (1µM) evoked contractions of endothelium-denuded superior mesenteric arterial strips. Trypsin (30 nM) failed to affect the contraction evoked by externally applied NA (Fig. 2A, B). Likewise, SL-NH2 (100µM) had no effect on the NA-induced contraction (Fig. 2C, D). For comparison, we investigated the effects of thrombin, a potent PAR-1 agonist on EFS-induced contraction. Thrombin (30 nM), did not show any effects on EFS-evoked contraction in endothelium-denuded superior mesenteric arterial strips (Supplmental Fig. 2). These results suggest that EFS-evoked contraction is suppressed not by PAR-1, but by PAR-2 activation and that the suppression by PAR-2 activation, may be mediated by inhibiting the peripheral sympathetic neuronal output that regulates peripheral vascular sympathetic tone. These results suggest that EFS-evoked contraction is suppressed by PAR-2 activation and that the suppression by PAR-2 activation may be mediated by inhibiting the peripheral sympathetic neuronal output that regulates peripheral vascular sympathetic tone.
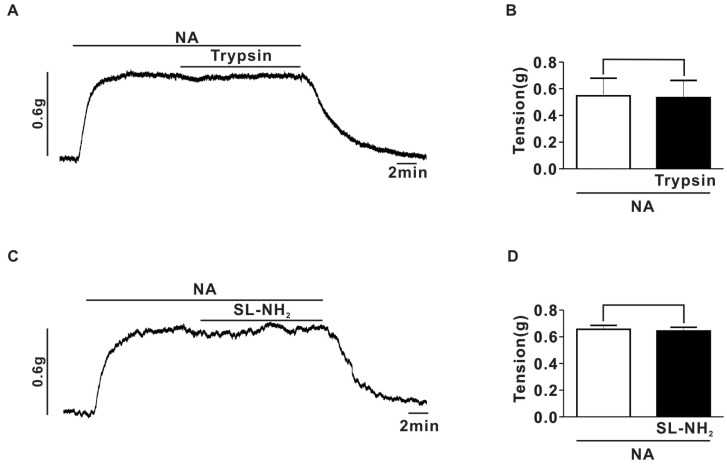
Fig. 2
Effect of NA and of NA plus PAR-2 agonists on the response to EFS-induced contraction in endothelium-denuded superior mesenteric arterial strip. (A) A representative trace of trypsin (30 nM) effect on the contraction evoked by externally applied NA (1µM). (B) Summary of trypsin effect on NA-evoked vasoconstriction (0.55±0.13 g for NA, 0.53±0.13 g for NA+trypsin, n=5, p>0.05). (C) A representative trace of SL-NH2 (100µM) effect on the contraction evoked by externally applied NA (1µM). (D) Summary of SL-NH2 effect on NA-evoked vasoconstriction (0.7±0.05 g for NA; 0.66±0.03 g for NA+SL-NH2, n=5, p>0.05).
![]()
The inhibitory effect of PAR-2 agonists on EFS-evoked contraction is mediated by ICa-N
The SCG neurons contribute to regulating the peripheral vascular sympathetic tone of superior mesenteric artery [26]. We first performed immunoblotting to confirm the presence of PAR-2 protein in SCG neurons. Consistent with previous data [27], expression of PAR-2 protein, with a predicted weight of 52 kDa, was identified in rat lung tissue, but was not detected in rat liver tissue. In addition, immunoreactivity of PAR-2 protein was also detected, indicating the presence of PAR-2 protein in rat SCG neurons (Supplmental Fig. 3).
To determine whether ICa-N are involved in the inhibition of EFS-evoked contraction by PAR-2 agonists, we examined the effect of ω-CgTx on inhibition of EFS-evoked contractions by PAR-2 agonists. Similar to the experiments in Supplmental Fig. 1C, ω-CgTx (1µM) suppressed EFS-evoked contraction in superior mesenteric arterial strips (Fig. 3). The pretreatment with ω-CgTx (1µM) nearly completely occluded the inhibitory effect of trypsin on EFS-evoked contraction (Fig. 3A and B). Similarly, SL-NH2 did not affect EFS-evoked contraction in the presence of 1µM ω-CgTx (Fig. 3C, D), suggesting that inhibition of EFS-evoked contraction by PAR-2 activation may be due to the modulation of ICa-N located in peripheral sympathetic nerve terminals.
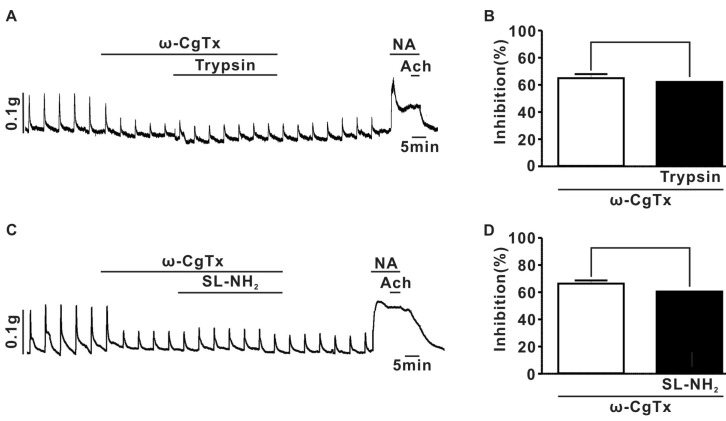
Fig. 3
Effects of ω-CgTx on inhibitory effect of PAR-2 agonists on EFS-induced contraction in rat endothelium-denuded superior mesenteric arterial strips. (A) A representative trace of the effects induced by consecutive application of ω-CgTx (1µM) and trypsin (30 nM) on EFS-induced contraction. (B) Summary of ω-CgTx effect on trypsin-induced inhibition of EFS-induced contraction (ω-CgTx; 65.0±2.9%, ω-CgTx+trypsin; 62.2±3.2%, p>0.05, n=5). (C) A representative trace of the effects induced by consecutive application of ω-CgTx (1µM) and SL-NH2 (100µM) on EFS-induced contraction. (D) Summary of ω-CgTx effect on SL-NH2-induced inhibition of EFS-induced contraction (ω-CgTx; 66.4±2.3%, ω-CgTx +SL-NH2; 60.6±2.3%, p>0.05, n=5).
![]()
PAR-2 agonists suppress the EFS-evoked overflow of NA in endothelium-denuded rat superior mesenteric arterial strips
We examined the effects of PAR-2 agonists on the EFS-evoked overflow of NA in superior mesenteric arterial strips. As shown in Fig. 4A, 30 nM trypsin reduced the EFS-evoked overflow of NA by 14.9±1.9%. However, 30 nM boiled trypsin (BT) did not have any effect on the EFS-evoked overflow of NA in superior mesenteric arterial strips (Fig. 4A). Likewise, 100 µM SL-NH2 decreased the EFS-evoked overflow of NA by 13.2±1.2% (Fig. 4A). On the contrary, 100µM LR-NH2, a control reverse peptide, had no influence on the EFS-evoked overflow of NA in superior mesenteric arterial strips (Fig. 4A).
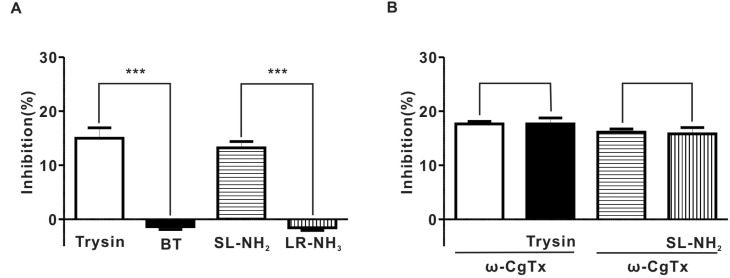
Fig. 4
Effect of PAR-2 agonist on EFS-evoked overflow of NA in endothelium-denuded rat superior mesenteric arterial strips. (A) summary of the effects of trypsin (7.1±0.2 pq/ml for control, 6.0±0.3 pq/ml for trypsin, n=4, p<0.001), SL-NH2 (7.0±0.3 pq/ml for control; 6.1±0.2 pq/ml for SL-NH2, n=4, p<0.001), BT (6.2±0.2 pq/ml for control, 6.3±0.2 pq/ml for BT, n=4, P>0.05), or LR-NH2 (6.7±0.2 pq/ml for control; 6.8±0.2 pq/ml for LR-NH2, n=4, p>0.05) on the EFS-evoked overflow of NA in rat superior mesenteric arterial strips. (B) summary of ω-CgTx effect on trypsin or SL-NH2-induced inhibition of EFS-evoked overflow of NA in rat superior mesenteric arterial strips (ω-CgTx; 17.6±0.5%, ω-CgTx+trypsin; 17.7±1.1%, p>0.05, n=4), (ω-CgTx; 16.1±0.6%, ω-CgTx+SL-NH2; 15.8±1.2%, p>0.05, n=4).
![]()
To determine whether ICa-N are involved in the inhibition of EFS-evoked overflow of NA by PAR-2 agonists, we examined the effect of ω-CgTx on inhibition of EFS-evoked overflow of NA by PAR-2 agonists. ω-CgTx (1µM) suppressed EFS-evoked overflow of NA by 16.9±0.4% (6.7±0.1 pq/ml for control; 5.6±0.1 pq/ml for ω-CgTx, n= 8, p<0.001) (Fig. 4B). The pretreatment with ω-CgTx (1µM) nearly completely occluded the inhibitory effect of trypsin on EFS-evoked overflow of NA (Fig. 4B). Similarly, SL-NH2 did not affect EFS-evoked overflow of NA in the presence of 1µM ω-CgTx (Fig. 4B).
PAR-2 agonists inhibit ICa in rat SCG neurons
To determine the relationship between PAR-2 and N-type voltage-gated Ca2+ channels directly, we examined the effects of PAR-2 agonists on ICa using the conventional voltage-clamp method in dissociated SCG neurons. To measure ICa in rat SCG neurons, the current was evoked by 200 ms depolarizing step pulses to a test potential of 0 mV from a holding potential of -80 mV. The average membrane capacitance of the CG neurons was 30.6±0.3 pF (n=56). Fig. 5A shows a typical example of the effect of trypsin on the ICa in SCG neurons. Application of 30 nM trypsin diminished ICa by 45.5±1.7% in an irreversible manner (Fig. 5A and C). Likewise, 100µM SL-NH2 also decreased ICa by 43.6±2.2% in an irreversible (Fig. 5B and C). BT (30 nM) (Fig. 5A and C) did not affect the ICa (inhibition 5.6±1.9%). Similarly, 100µM LR-NH2 (Fig. 5B and C) did not affect ICa (inhibition 8.0±2.8%).
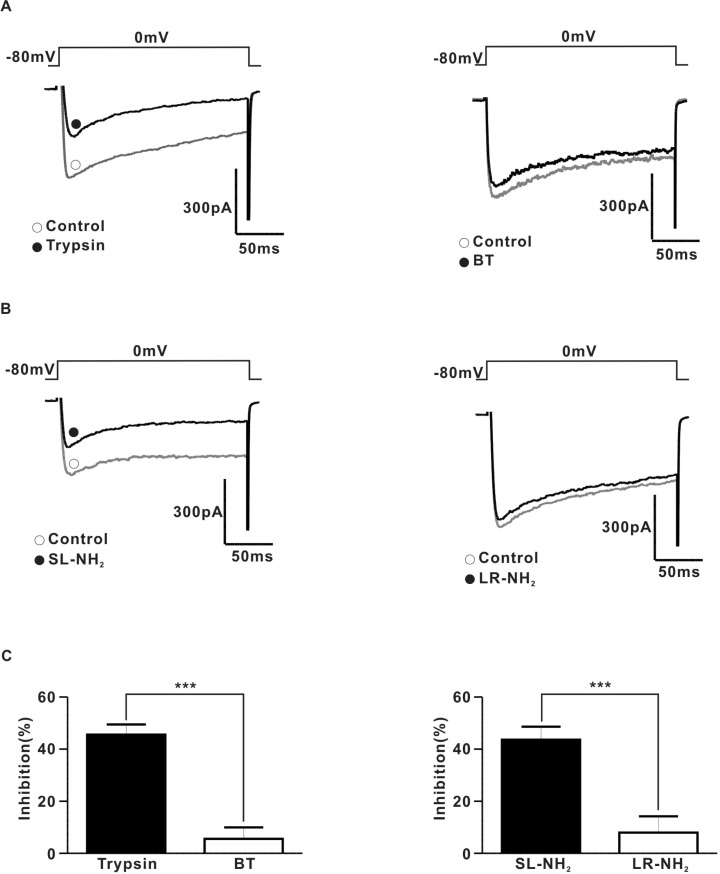
Fig. 5
PAR-2 agonists inhibited ICa in rat SCG neurons (A) Left, a representative trace of ICa in the presence (●) and absence (○) of 30 nM typsin. Right, a representative trace of ICa in the presence (●) and absence (○) of 30 nM BT. (B) Left, a representative traces of ICa in the presence (●) and absence (○) of 100µM SL-NH2. Right, a representative trace of ICa in the presence (●) and absence (○) of 100µM LR-NH2. (C) left, a summary of ICa inhibition by trypsin (815.5±104.9 pA for control; 428.0±54.1 pA for trypsin, n=5) and BT (906.6±115.7 pA for control, 863.1±125.6 pA for BT, n=5, p>0.05). Right a summary of ICa inhibition by SL-NH2 (902.5±111.6 pA for control, 506.1±58.8 pA for SL-NH2, n=5) or LR-NH2 (786.8±67.1 pA for control, 727.1±71.2 pA for LR-NH2, n=5, p>0.01).
![]()
DISCUSSION
The present study demonstrates that PAR-2 agonists suppress neurogenic contraction evoked by EFS by modulating peripheral sympathetic tone, which regulates peripheral vascular contractility in endothelium-denuded superior mesenteric arterial strips. We also found that the suppression of peripheral sympathetic tone by PAR-2 activation is mediated mainly by modulating ICa-N located in sympathetic nerve terminals.
Since Al-Ani et al. [2] first reported that PAR-2 agonists caused an endothelium-dependent relaxation in rat aortic rings in nitric oxide (NO)-dependent manner, PAR-2 activation has been reported to play an important role in regulating blood pressure in various vascular tissues such as porcine coronary arteries [28,29], rat femoral artery [6,30], rat pulmonary arteries [30] and basilar arteries [30,31]. However, in all of these cases, including resistance vessels, PAR-2-induced vasorelaxation was partially maintained even after inhibition of NO synthase by L-NAME, suggesting the involvement of another mechanism, distinct from the L-Arginine/NO pathway in PAR-2-induced hypotension. Consistent with in vitro data, it has been seen with in vivo preparations that PAR-2 agonist peptides applied by intravenous injection, causes hypotension in anesthetized rats [6,7] and mice [9,10]. This PAR-2-induced hypotension is also partially inhibited by L-NAME, suggesting the involvement of an NO-independent mechanism [6,8]. In addition, this hypotension is not affected by alterations in kidney or heart function, suggesting an effect of the PAR-2 agonist peptides on the peripheral vascular system [6,9]. These results therefore suggest that another mechanism, other than the NO pathway in endothelium, may be involved in hypotension induced by PAR-2 activation.
Neural regulation of blood pressure is accomplished by regulating peripheral vascular resistance through the peripheral sympathetic nervous system. The postganglionic fibers of the peripheral sympathetic nervous system innervate the arteries and arterioles, regulating peripheral vascular contraction. NA release from sympathetic nerve terminals following peripheral sympathetic activation increases the resistance of peripheral blood vessels via vasoconstriction of vascular smooth muscle. In normal physiological states, the peripheral sympathetic outputs to blood vessels are tonically active and therefore any substance, which can alter peripheral sympathetic activity, can readily regulate peripheral vascular resistance, which in turn affects blood pressure. So, it is possible that PAR-2 activation may regulate blood pressure by modulating activity of peripheral sympathetic neurons which innervate peripheral resistant vasculature. In addition, PAR-2-induced hypotension is quite sustained, lasting about 2~3 min, compared to the duration of PAR-1-induced hypotension [6,8,9] and the duration of PAR-2-induced hypotension is significantly prolonged by ganglion-blockage with chlorisondamine [8], suggesting that the regulation of peripheral sympathetic activity by PAR-2 activation, may be involved in PAR-2-induced hypotension.
In the present study, PAR-2 agonists suppressed neurogenic contractions evoked by EFS, which mimics vasoconstriction mediated by peripheral sympathetic activation in endothelium-denuded superior mesenteric arterial strips. The PAR-2 agonists did not affect contraction elicited by the external application of NA. On the contrary, thrombin, a potent PAR-1 agonist, had no effect on EFS-evoked contraction in endothelium-denuded superior mesenteric arterial strips, suggesting a lack of peripheral sympathetic involvement in PAR-1-induced hypotension. These results may explain the difference of time course for between PAR-1 and PAR-2 mediated hypotension.
As for the pathphysiological function of PAR-2 on blood pressure, PAR-2 usually is activated by serine protease such as trypsin or trytase which is mainly released from endothelial cells and other cells during inflammation [32] or coagulation factor XI which is massively activated by tissue factor released by endotoxin in septic condition [33]. Therefore, PAR-2 is usually considered to be involved in regulation in blood pressure in pathological state. But, recently, McGuire et al., [34] reported that PAR knockout mice exhibited moderated elevation of systolic arterial and pulse pressure, suggesting PAR-2 involvement in blood pressure regulation in normal physiological states. But, further studies are needed to elucidate a detailed mechanism and functional role for PAR-2-induced hypotension in normal physiological states.
ICa-N play an important role in modulating peripheral sympathetic activity in postganglionic sympathetic neurons [14,15]. Increase in intracellular Ca2+ concentration due to Ca2+ influx through voltage-gated Ca2+ channels triggers NA release in peripheral sympathetic nerve terminals. Therefore, any substance that modulates Ca2+ channel activity can influence NA release and therefore regulate peripheral sympathetic tone [35]. Therefore, PAR-2-mediated inhibition of peripheral sympathetic output may be associated with ICa-N activity in sympathetic nerve terminals. In the present study, the blockade of ICa-N with ω-CgTx nearly completely occluded the suppression of EFS-evoked contraction by PAR-2 agonists. Consistent with patchclamp data, PAR-2 agonists suppressed the EFS-evoked overflow of NA in endothelium-denuded rat superior mesenteric arterial strips, which was nearly completely occluded by ω-CgTx. To determine the effect of PAR-2 agonists on N-type Ca2+ channels in the peripheral sympathetic neurons more directly, we tested the effect of PAR-2 agonists on ICa using voltage-clamp in rat SCG neurons which regulate peripheral sympathetic tone of the mesenteric vasculature. As shown in Fig. 5, PAR-2 agonists diminished ICa in an irreversible manner in rat SCG neurons.These results suggest that the suppression of peripheral sympathetic activity by PAR-2 agonists is mediated mainly by the inhibition of ICa-N located in peripheral sympathetic nerve terminals.
In conclusion, the present findings demonstrate that the activation of PAR-2 suppresses peripheral sympathetic outflow by modulating ICa-N activity located in peripheral sympathetic nerve terminals, which appear to be involved in PAR-2-induced hypotension. These results demonstrated, for the first time, a possible neuronal mechanism for PAR-2-induced hypotension and provide a basic and theoretical framework that could lead to the development of new agents for the treatment of hypertension.
 XML Download
XML Download