

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ischemic heart disease (IHD) is a major cause of mortality worldwide [1]. Most patients with myocardial infarction (MI) due to coronary artery disease (CAD), also have other risk factors including aging, hypertension, atherosclerosis, diabetes, and thyroid diseases that lead to increased incidence of MI [2]. Decreased coronary blood flow leads to reduction in delivery of blood, oxygen, and other nutrients to the heart, leading to heart ischemia [3]. Regardless of requirement of reperfusion to re-establish the normal function of the myocardium, sudden reperfusion of ischemic heart generates additional damage defined as ischemia-reperfusion (IR) injury [4,5].
The myocardial response to IR injury can be moderated via various interventions such as ischemic postconditioning (IPost) [6,7]. IPost, induced using short cyclic episodes of IR immediately at the beginning of reperfusion after prolonged ischemia [8,9], is feasible in clinical settings since it is applied immediately subsequent to the infarction incident [10]. Most studies on IPost have been carried out on normal animals; however CAD mostly co-exists with additional morbidities [1,2]. Before utilizing of IPost in a clinical setting, further research is needed to determine any protective effects it may have on disease associated with cardiovascular diseases [11].
Patients with overt or subclinical hyperthyroidism are at increased risk of cardiovascular morbidity and mortality [2]. Hyperthyroidism is commonly associated with increased heart rate, and high blood, and pulse pressure [12,13]. Serious tachycardia in the heart of hyperthyroid rats predispose the heart to reperfusion injury [13,14]; indeed, elevated thyroid hormone levels could have detrimental effects on its response to ischemia. Generation of reactive oxygen species (ROS) are increased in hyperthyroidism weakening the antioxidant defense system in heart [15].
Nitric oxide (NO) is produced by NO synthase enzymes and plays an essential role in cardiac functioning. Increased NO production following myocardial ischemia leads to IR injury. Conditions that escalate both NO and oxidative stress production also contribute to IR injury [14,16]. In this regard, no study has hitherto addressed the role of IPost in changes of NO content in hearts of hyperthyroid rats; neither has the effect of IPost on myocardial IR-injury in hyperthyroidism been investigated. The association of hyperthyroidism with cardioprotection by IPost remains unclarified.
Hence, the aim of this study was to investigate effect of IPost against IR injury in an in vitro model of T4-induced hyperthyroidism in male rats; the alterations of NO metabolites (NOx) following IR and IPost were also determined.
Go to : 
METHODS
Animals
In this study, 56 male Wistar rats (2-month old) were provided from laboratory animal house of the Research Institute for Endocrine Sciences (RIES), Shahid Beheshti University of Medical Sciences. All animals were housed in an animal room with temperature and light controlled (23±1℃ 12/12-h dark-light cycle) conditions, and had free access to food and water at all times before initiation of surgical procedures. This study was carried out according to guidelines for the care and use of laboratory animals confirmed by the RIES ethics committee. For IR experiments, the control and hyperthyroid animals were randomly divided into 7 subgroups (n=8 each): Control (C), control-IR (C-IR), control-postconditioning (C-IPost), hyperthyroidism (HP), hyperthyroidism-IR (HP-IR), hyperthyroidism- postconditioning (HP-IPost), and hyperthyroidism-postconditioning-aminoguanidine (HP-IPost-AG). Hyperthyroidism in rats was induced by adding T4 (Sigma; 12 mg/l) to the drinking water for a period of 21 days [17].
Serum thyroid hormone measurement and determination of treatment efficacy
Serum total T4 (thyroxine), T3 (triiodothyronine), and TSH (thyroid stimulating hormone) levels in the control and hyperthyroid animals were determined at the end of treatment phase using commercial Elisa kits. Changes in the heart weight (HW) to body weight ratio (cardiac hypertrophy) during the treatment and citrate synthase (CS) activity (indicator for oxidative capacity) in the soleus muscle of rats were assessed for determining efficacy of treatment with T4, the latter using the Srere procedure [18].
Isolated heart preparation
All animals were anesthetized with an intraperitoneal injection of combination of ketamine and xylazine (50 mg/kg and 10 mg/kg,). Perfusion experiments were carried out 48 h following the last T4 dose. The hearts of control and hyperthyroid rats were rapidly excised and embedded in an ice-cold perfusion buffer; the aorta was then cannulated and the hearts were fixed on the Langendorff perfusion device and perfused through the aorta with a Krebs-Henseleit solution containing (mM/L): NaCl 118, NaHCO3 25, KCl 4.7, MgCl2 1.2, CaCl2 2.5, KH2PO4 1.2, and glucose 11 at a constant pressure (75 mm Hg) and pH 7.4. The Krebs solution was gassed with a combination of 95% O2 and 5% CO2 at 37℃. All isolated hearts were stabilized for 20 minutes with the purpose of obtaining baseline data. After stabilization period of 20 min, in C-IR and HP-IR groups, hearts were subjected to global ischemia for 30 minutes followed by reperfusion for 120 minutes. The hearts from rats in the HP-IPost-AG group were perfused during the last 10 min of stabilization with 100 µmol/L of AG (Sigma-Aldrich), a selective iNOS-inhibitor [19]. IPost was induced by six cycles of 10 sec reperfusion and ischemia (6 cycles of 10s IR) immediately at the onset of reperfusion following 30 minutes of global ischemia. Left ventricular hemodynamical parameters were measured via a Latex balloon inserted in the left ventricle. The balloon capacity was adjusted to create 5~10 mm Hg of end diastolic pressure in all hearts by filling it with water. Hemodynamic parameters were digitized and recorded by a data acquisition system (power lab, AD instrument, Australia). Left ventricular end diastolic pressure (LVEDP), heart rate (HR), left ventricular developed pressure (LVDP), and the maximum rate of increase and decrease of left ventricular pressure (±dp/dt) were recorded. Postischemic recoveries were assessed by LVDP, LVEDP, and ±dp/dt and are represented as percent of the basal values.
Measurement of NOx
Following 2-hour reperfusion, samples of left ventricle (LV) tissue were immediately frozen in liquid nitrogen and stored at -80℃. NOx contents in serum and myocardium homogenates were determined using the Griess method [20]. Briefly, after homogenization of samples in PBS (phosphate-buffered saline) (1:5, w/v), the homogenates were centrifuged at 15,000 g for 20 min at 4℃. The supernatants were removed from the homogenates and were deproteinized by addition of zinc sulfate (15 mg/ml). Serum samples were also deproteinized using zinc sulfate (15 mg/ml) and centrifuged at 10,000 g for 10 min at 4℃. For both serum and tissue samples, a 100 µl of the supernatant was added to a microplate well and 100 µl vanadium (III) chloride (8 mg/ml) was added to each well (for reduction of nitrate to nitrite), followed by addition of 50 µl sulfanilamide (2%) and 50 µl NEDD (N-(1-naphtyl) ethylendiamine dihydrochloride) (0.1%). After 30 min incubation at 37℃, the absorbance was read at 540 nm using the ELISA reader (BioTek, Powerwave XS2). NOx concentrations in serum and heart homogenates samples were measured from the linear standard curve established by 0-150 and 0-50 µmol/L of sodium nitrate, respectively. Heart and serum NOx levels are presented as µmol/L. Intra-assay coefficient of variation was 5.2%.
Measurement of infarct size
At the end of the reperfusion period, infarct sizes (IS) were determined as described previously [21]. The frozen heart samples were cut into thin slices (2~3 mm) and were incubated for 10 min in 1% of 2, 3, 5-triphenyltetrazolium chloride (TTC) in phosphate buffer solution 20 mM/L, pH 7.4 at 37℃. The slices were immersed in 10% formalin for 24 h to identify viable myocardium as red stained, while necrotic (infracted) tissue remains unstained (pale gray). The sections were photographed using a digital camera version DV101 (Samsung, Japan). IS was measured by Photoshop CS6 software (version 13) and expressed as percentage of the total area.
Statistical analyses
All data are presented as means±SEM. LVDP, LVEDP, ±dp/dt, IS and NOx values were evaluated by repeated measure ANOVA; a post-hoc Tukey test was applied using SPSS software for comparing means between groups, with p values <0.05 being considered significant.
Go to :
RESULTS
CS activity in soleus muscle and thyroid hormone levels (T3, T4) in serum were significantly higher, whereas serum TSH was significantly lower in hyperthyroid rats. In addition, body weight changes in hyperthyroid rats were not significantly different from controls, and ratio of heart weight to body weight was higher in hyperthyroid rats (Table 1).
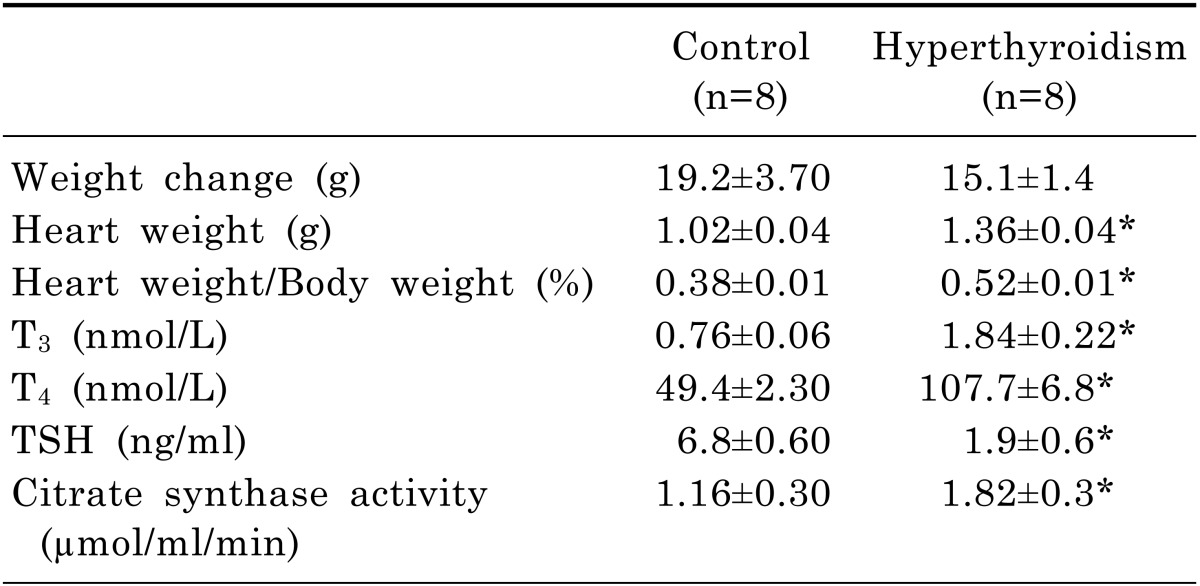
Preischemic hemodynamic values were significantly lower in the hyperthyroid groups than controls (Table 2). When ischemia was induced by the stopping the inflow of the perfusion solution, the LVDP, ±dp/dt, and heart rate rapidly declined and ceased in the isolated hearts
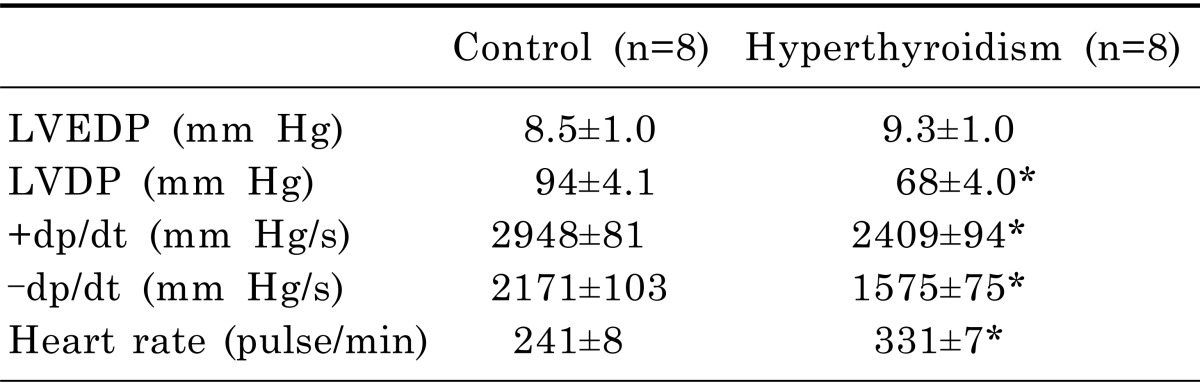
Post-ischemic ±dp/dt and LVDP decreased significantly in the HP-IR group, compared with the C-IR group following ischemia (30 min) and reperfusion (120 min). IPost significantly improved the ±dp/dt and LVDP in reperfusion phase in the C-IPost group; however, it did not improve ±dp/dt and LVDP in the HP-IPost group. Following IR, post-ischemic ±dp/dt and LVDP increased significantly in the HP-IPost-AG group, compared to the HP-IPost group (Fig. 1).
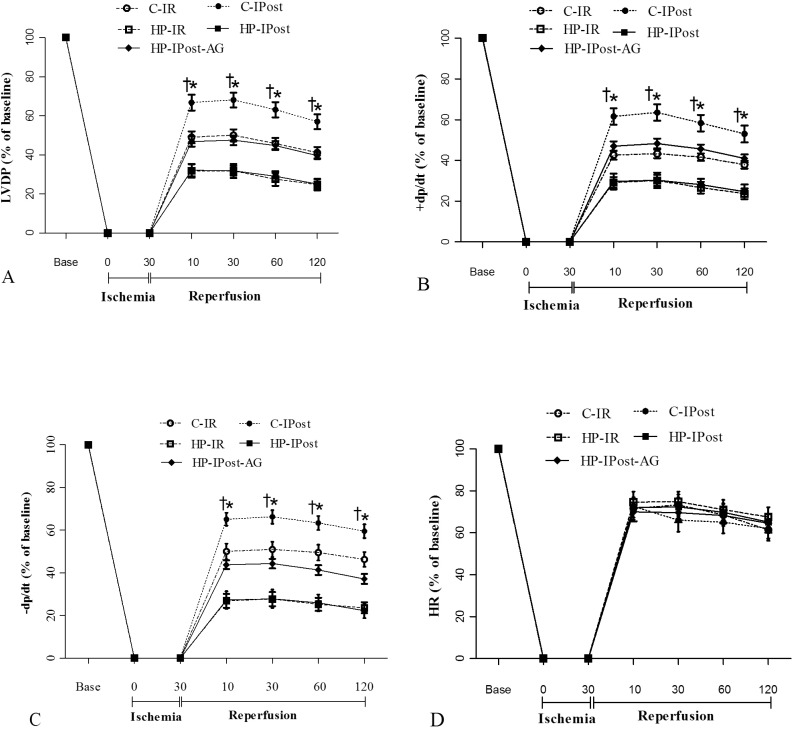 | Fig. 1Alterations of cardiac function during reperfusion; (A) Left ventricular developed pressure (LVDP); (B) Maximum increase in left ventricular pressure (+dp/dt); (C) Decrease in left ventricular pressure (-dp/dt); (D) Heart rate; control-ischemia reperfusion (C-IR), C-ischemic postconditioning (C-IPost), hyperthyroid-IR (HP-IR); hyperthyroid-IPost (HP-IPost), and hyperthyroid-IPost-aminoguanidine (HP-IPost-AG); data are mean±SE (n=8 rats); *p<0.05 significant difference between C-IR and C-IPost groups. †p<0.05 significant difference between HP-IPost and HP- IPost-AG.
|
During the 30 minutes ischemia, hyperthyroid group showed a significant increase in LVEDP (contracture), compared to controls. IPost significantly reduced the LVEDP during reperfusion phase in the C-IPost group; in addition, IPost in combination with AG, significantly reduced the LVEDP during reperfusion phase in the HP-IPost-AG group compared to HP-IPost (Fig. 2).
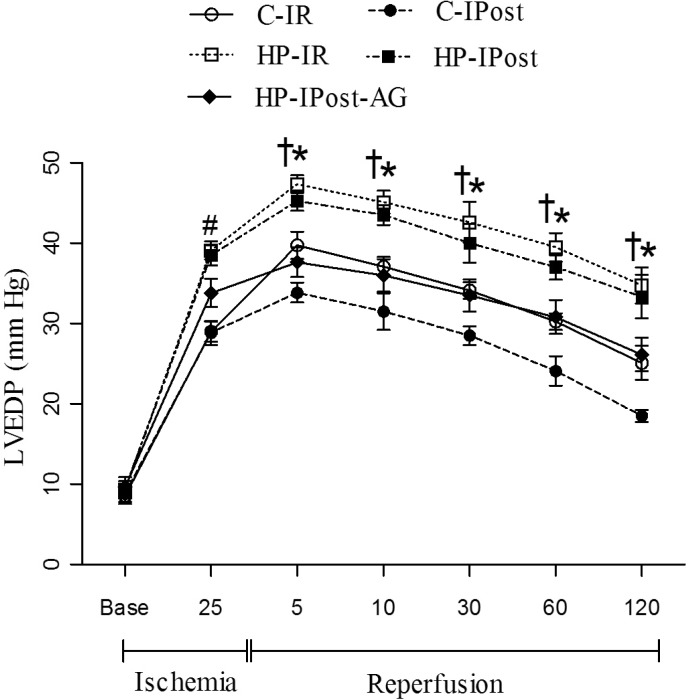 | Fig. 2Alterations of LVEDP during IR; left ventricular end diastolic pressure (LVEDP); control-ischemia reperfusion (C-IR), C-ischemic postconditioning (C-IPost), hyperthyroid-IR (HP-IR); hyperthyroid-IPost (HP-IPost) and hyperthyroid-IPost-aminoguanidine (HP-IPost-AG); data are mean±SE (n=8 rats); *p<0.05 significant difference between C-IR and C-IPost. #p<0.05 significant difference between C-IR and HP-IR, and between C-IPost and HP-IPost. †p<0.05 significant difference between with HP-IPost and HP-IPost-AG.
|
Heart and serum NOx levels were significantly higher in the hyperthyroid group, compared to the controls. The level of NOx was enhanced significantly in both groups of HP-IR and HP-IPost. IPost had no significant effect on reduction of heart NOx level in the HP-IPost group, although, IPost significantly reduced the IR-induced enhancement in heart NOx of the control group. AG significantly reduced heart NOx levels in hyperthyroid rats subjected to IPost (Fig. 3). There was significant difference in the IS between groups of C-IR and HP-IR animals (47.5±1.9% vs. 64±2.3%). IPost significantly reduced the IS in control group, while it had no effect in the hyperthyroid group; IPost in presence of AG significantly reduced the IS in HP-IPost-AG group (60.5±2.04 vs. 47.60±1.72) (Fig. 4).
 | Fig. 3The alterations of NOx in heart of control and hyperthyroid groups; control- ischemia reperfusion (C-IR); C-ischemic postconditioning (C-IPost); hyperthyroid-IR (HP-IR); hyperthyroid-IPost (HP-IPost), and hyperthyroid-IPost-aminoguanidine (HP-IPost-AG); data are as mean±SE (n=8 rats); *p<0.05 compared with control group. **p<0.05 compared with C-IR group. #p<0.05 compared with hyperthyroid group. †p<0.05 compared with HP-IPost group.
|
 | Fig. 4The alterations of infarct size in heart of control and hyperthyroid groups. control- ischemia reperfusion (C-IR); C-ischemic postconditioning (C-IPost); hyperthyroid-IR (HP-IR); hyperthyroid-IPost (HP-IPost), and hyperthyroid-IPost-aminoguanidine (HP-IPost-AG); data are mean±SE; (n=6 rats) as present of total area; *p<0.05 compared with control group. **p<0.05 compared with C-IR group. #p<0.05 compared with hyperthyroid group. †p<0.05 compared with C-IR group. ††p<0.05 compared with HP-IPost group.
|
Go to :
DISCUSSION
Our findings indicate that hyperthyroidism increase injury induced by IR in rat heart, which may be due to increasing of NO production. IPost provides protective effects against IR injury in control rats, but, has no effect in hyperthyroid rats. Heart hypertrophy increases both the soleus muscle CS activity, and also levels of serum thyroid hormones (T3 and T4), while decreasing TSH levels, showing that hyperthyroidism has been effectively induced.
In our study, preischemic values of LVDP and ±dp/dt were lower in the hyperthyroid rats; the hearts from hyperthyroid rats, exhibited a decreased recovery of LVDP and ±dp/dt following IR indicating that these hearts are vulnerable to IR injury, findings consistent with previous studies [13,14,15]. It has been reported that chronic T4 administration (21 days) results in increased calcium concentration in cardiac cells [22], which, in turn, causes mitochondrial malfunction, and, consequently cell death [22]. Contradictory to our results, which were on a long-term basis it has been reported that hyperthyroidism provides cardioprotective effects following IR [23,24]; a likely explanation for this difference may be moderately short-term T4 administration used in these studies [24].
In this study, pre-ischemic heart rate was higher in the hyperthyroid group than the controls; in this regard, studies have been shown that hyperthyroidism increseas the heart rate through various mechanisms, such as augmented L-type calcium channels [25], elevated activity of Ca+2 ATPase [26], and declined phospholamban [27]. Our results show that hearts from hyperthyroid rats subjected to global ischemia developed ischemic contracture (increased LVEDP); it has been reported that ATP depletion and increasing Ca2+ concentration in heart cells are crucial factors in ischemic contracture [28]. High levels of thyroid hormones can increase the susceptibility of these hearts to IR and may be associated with H2O2 production and mitochondrial dysfunction [15,29,30].
In our study, IPost failed to offer any protection in hearts of hyperthyroid rats, unlike the hearts of control rats, in which it seems that IPost had a cardioprotective effect, via inhibiting mPTP (mitochondrial Permeability Transition Pore). It has been identified that in hyperthyroid status during myocardial reperfusion injury, opening of mPTP occurs possibly due to ROS production and mitochondrial Ca2+ overload [31,32]. Therefore, it seems that IPost possibly loses its effectiveness in a hyperthyroid group, a finding similar to those from diabetic rats, where preconditioning and IPost cannot protect the hearts of diabetic rats against IR injury [33,34].
In our study, IPost showed cardioprotective effect in non-hyperthyroid animals by reducing the IS, but failed to reduce IS in hyperthyroid animals. Similar to our results, it has been reported that cardioprotective effects of IPost in reducing IS abrogated in disease state including metabolic syndrome [35] and diabetes [34,36].
In the current study, hypertrophy occurred along with increasing of heart NOx metabolites in the hyperthyroid group; it has been reported that thyroid hormones lead to activation of the inducible NO synthase (iNOS) directly and increasing NO levels [37]. In the hearts of hyperthyroid rats, upregulation of iNOS causes NO overproduction, which is accompanied by elevated oxidative stress [37,38]. Also, other studies have shown that the high production of NO may contribute in cardiac hypertrophy [39,40]. Similar to our results, increasing of heart NO level in hyperthyroid rats following IR has been previously reported [14,30]. Our results show that hyperthyroidism increases injuries induced by IR in rat hearts, which is possibly due to elevated NO levels. It has been hypothesized that hyperthyroidism could lead to reperfusion injury by increase of NO production, which, in turn increases nitro-oxidative stress [30]. Data shows that the high levels of NOx during the IR stage is possibly an important factor of hyperthyroid-induced cardiomyopathy [14]. The interaction between both NO and free radicals has been shown to increase lipid peroxidation in reperfusion [30]. Thus overproduction of free radicals in hyperthyroidism is associated with reduced antioxidant defense. The studies have shown that the harmful effect of NO on IR is mediated by peroxynitrite [41]. The combination of high level of NO with superoxide can produce peroxynitrite that this is considered a toxic agent in the death of myocardial cells [41,42].
In this study, IPost, significantly diminished NOx in the hearts of control rats, whereas, we observed increasing of NOx in the HP-IPost group. In addition, IPost did not protect the heart from IR injury in hyperthyroid rats. Some investigations have demonstrated that IPost protects the non-diseased rat heart from IR injury, by diminishing of the NO concentration in the heart, subsequent to ischemia phase [41,42,43,44]. Therefore, it seems that the increasing of NO and free radicals in hyperthyroidism, causes loss of IPost efficiency in the hearts of hyperthyroid rats; this pattern that NO overproduction by iNOS which prevents cardioprotective effect of IPost in hyperthyroidism has also been reported in diabetic rats [45,46]. It has been shown that iNOS inhibition could restore cardioprotective effects of IPost in diabetes [46]. Similarly in the current study, we showed that IPost in presence of AG, which selectively inhibits iNOS, provide cardioprotection in hyperthyroid rats.
In conclusion, hyperthyroidism increased susceptibility of heart to IR injury and unlike control rats, IPost could not provide protection against this injury due to NO overproduction; cardioprotective effects of IPost in hyperthyroid rats are restored in presence of iNOS inhibition.
Go to :
 XML Download
XML Download