

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
AD
Alzheimer's disease
Aβ
amyloid beta
Aβ1-42
amyloid beta 1-42
NFT
neurofibrillary tangle
CSF
cerebrospinal fluid
MCI
mild cognitive impairment
NMDA
N-methyl-D-aspartate
FDA
Food & Drug Administration
APP
amyloid precursor protein
Abo
amyloid beta oligomer
PPAR-g
peroxisome proliferator-activated receptor gamma
BBB
blood-brain-barrier
RAGE
receptor for advanced glycation end products
LRP-1
low-density lipoprotein receptor-related protein 1
PET
positron emission tomography
IVIG
intravenous immunoglobulin
GSK-3
glycogen synthase kinase 3
ADNI
Alzheimer's Disease Neuroimaging Initiative
MRI
magnetic resonance imaging
FDG
2-[18F]-fluoro-2-deoxy-D-glucose
AICD
C-terminal fragment of amyloid precursor protein
INTRODUCTION
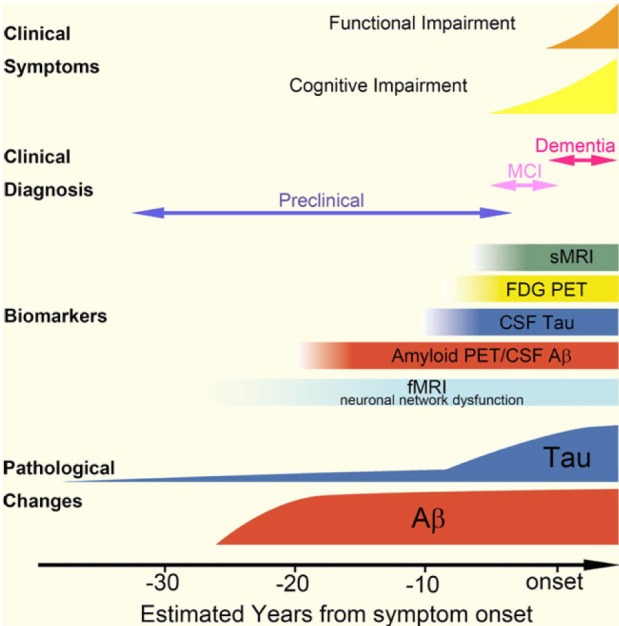
Alzheimer's disease (AD), a progressive neurodegenerative disorder, is the most common form of irreversible dementia, and it carries with it a considerable human, social and economic burden. Following the onset of pathological changes in the brain that include the progressive accumulation in the CNS of amyloid beta (Aβ) deposits and neurofibrillary tangles (NFTs) formed by pathological tau, which is thought to begin more than 15 (Aβ) to 10 (NFTs) years before cognitive impairments become clinically manifest, AD patients primarily develop progressive deterioration of episodic memory and a global decline in their cognitive functions. Among several mechanistic and pathological substrates that contribute to the gradual progression of AD over time, the major neuropathological substrates of AD are the aggregation and accumulation of misfolded Aβ and the intracellular deposition of fibrillized and hyperphosphorylated tau proteins (Fig. 1).
Currently, the greatest utility for biochemical cerebrospinal (CSF) biomarkers of AD may be for the early and more reliable diagnosis of AD, which places measures of the levels of CSF Aβ1-42 and tau proteins (total tau and tau phosphorylated at Thr181) in the revised version of diagnostic criteria for AD research [1,2,3,4]. Thus, abundant data from numerous studies carried out in centers across the globe over the past 20 years shows that the levels of Aβ1-42 in CSF of AD patients are significantly lower than in age-matched healthy elderly controls, whereas the levels of total tau (t-tau) and tau phosphorylated at threonine 181 (p-tau181) in AD CSF are significantly higher than those of controls. Moreover, it has been suggested that these CSF biomarkers are useful to differentiate those mild cognitive impairment (MCI) patients who progress to develop AD within the subsequent several years from the stable MCI patients [5,6,7,8,9,10].
The current standard pharmacotherapy for cognitive improvement in AD patients includes acetylcholinesterase inhibitors and the N-methyl-D-aspartate (NMDA) antagonist memantine. However, the approval of these drugs has not been based on their ability to slow disease progression but to improve the clinical symptomatology. Hence, only symptomatic drugs with transient benefits have been approved for clinical use in AD patients by the US Food and Drug Administration (FDA). Given the fact that genetic and pathological evidence strongly supports the "amyloid cascade hypothesis", several strategies of reducing Aβ accumulation in the brain have been applied to develop "disease-modifying therapy" to cure AD. In addition, a correlation between cognitive dysfunction and neurofibrillary tangle load that is more robust than that between Aβ load and cognitive impairments has led to a parallel strategy to develop tau focused therapies that inhibit and/or block tau aggregate formation, promote the clearance of tau pathology or correct for the loss of tau function when tau is sequestered in NFTs [11]. Other strategies for modifying disease progression include anti-inflammation, metabolic approaches, neurotrophin-based approaches and mitochondrial targets. It has been estimated that a disease-modifying therapy that could delay both dementia onset and progression by one year would reduce the prevalence by 9.2 million cases of the disease by the year 2050 [12,13]. To reduce the number of future AD cases, a variety of attempts to develop a drug that provides a disease-modifying effect against probable AD have been made; however, there are no approved disease-modifying therapies for AD at this time. Based on lessons from previous clinical trials in probable AD patients, there is a growing consensus that initiation of disease-modifying treatment before the onset or early phase of the disease (before the onset of clinical dementia) may be necessary. To maximize the power of clinical trials in patients without clinical dementia, valid biomarkers for the early detection of patients with AD pathology or the prediction of patients presenting a prodromal phase of AD (commonly referred to as MCI), who will likely develop AD in the future, would be helpful. Currently, Aβ1-42 and tau proteins (t-tau and p-tau181) in CSF are the most reliable biochemical biomarkers for the early detection of AD, the differentiation of AD from other forms of dementia, and the prediction of MCI progression to AD. In this review, the current development of AD therapeutics, particularly those drugs targeting amyloid and tau pathology, and the clinical performance of CSF biomarkers for AD diagnosis are summarized. The advantages and unresolved issues of CSF biomarkers in clinical trial design are also discussed.
TARGETS OF DEVELOPING DRUGS IN AD PATHOPHYSIOLOGY
Target related to amyloid production, aggregation and clearance
The molecular chain of events that ultimately results in synaptic and neuronal loss in the brain is complex and remains largely unresolved, even in the well-known pathologic hallmarks of AD, i.e. the deposits of Aβ peptides in amyloid plaques and other types of aggregates and NFTs formed by misfiled and fibrillized tau. A widely accepted hypothesis is that AD might be initiated by the abnormal (amyloidogenic) processing of amyloid precursor protein (APP) followed by the aggregation and accumulation of Aβ in the brain. The strategy to develop anti-Aβdrugs targeting the canonical amyloid cascade can be classified according to the mechanism of action: reduction of Aβ1-42 production, prevention of Aβ oligomer (Aβo) formation, and acceleration of Aβ clearance (Fig. 2). Based on the amyloid hypothesis, several drugs to reduce Aβ burden are being developed for patients with mild-to-moderate AD.
Considering that genetic mutations in the APP or PSEN1 genes cause familial AD and that β-secretase (β-site APP cleaving enzyme, BACE1) knock-out mice showed drastically reduced Aβ levels in the brain, the inhibition of β- and/or γ-secretases can be a strategy to block the initiation of the amyloid cascade. Several first-generation γ-secretase inhibitors (e.g., semagacestat) were tested in clinical studies; however, because γ-secretase is also involved in the processing of Notch, a Phase 3 trial for semagacestat in patients with mild-to-moderate AD not only failed to achieve its predetermined end points, but also worsened clinical measures and increased the incidence of skin cancer [14]. Other Notch-sparing, second-generation γ-secretase inhibitors (e.g., begacestat, avagacestat, PF-3804 014 and NIC5-15) are now in the early phase of clinical trials [15,16,17]. Because amyloidogenic Aβ species are generated by sequential activation of β-secretase and γ-secretase, the inhibition of β-secretase can be the second strategy to suppress the amyloidogenic pathway. However, β-secretase has many endogenous substrates that are not related to APP processing; therefore, no Phase 3 clinical trials of new β-secretase inhibitors are developing, but anti-β-secretase antibodies and the oral compound CTS- 21166 are under investigation. Interestingly, previous research found that the thiazolidinedione antidiabetic drugs (e.g., rosiglitazone and pioglitazone) are potentially beneficial in that they appear to suppress Aβ burden via peroxisome proliferator-activated receptor γ (PPAR-γ) activation [18,19]. PPAR-γ activators can suppress β-secretase expression and accelerate APP degradation by increasing its ubiquitination [20]. Although one clinical trial evaluating rosiglitazone demonstrated a beneficial effect on cognition, particularly in ApoE4 negative patients [21], the larger confirmatory clinical trials failed to demonstrate a beneficial effect on cognition in AD patients [22,23,24]. In addition, a recent warning by the US FDA about possible cardiac risks associated with rosiglitazone and the negative preliminary results led to a discontinuation of the further development of the rosiglitazone program for AD. There is a conflicting result regarding efficacy in AD for pioglitazone, another PPAR-γ activator [25,26]. Another strategy to reduce pathogenic Aβ production is the upregulation of α-secretase activity, leading to upregulation of neuroprotective sAPPα secretion. Several drugs have been tested in early phase clinical trials, but the results are not yet available.
Aβ, particularly Aβ1-42, is prone to aggregation and forms toxic Aβo. Given the evidence that the neurotoxic potency of Aβo is higher than the Aβ monomer or insoluble Aβ amyloid fibrils [27,28], compounds inhibiting Aβ aggregation or destabilizing Aβo seem to be promising drug candidates for AD. The initial anti-aggregant is tramiprosate (homotaurine), which binds preferentially to soluble Aβ; however, the outcome of a Phase 3 trial was not significant [29]. Another anti-aggregant that inhibits metal-induced Aβ oligomerization, PBT2, promotes Aβo clearance and improves cognition in an animal model [30]. In a Phase 2a clinical trial in patients with mild AD for 12 weeks, PBT2 was well-tolerated, reduced CSF Aβ1-42 concentrations and improved executive function [31]. However, recent news releases from Prana reported that PBT2 failed to meet its primary endopoint of reducing Aβ plaques in a 12-month phase 2 "IMAGINE" trial (http://pranabio.com/news/prana-biotechnology-announces-top-line-results-phase-2-imagine-trial-pbt2-alzheimers-disease/#.U4Ol1WdOVaQ). Based on the in vitro and in vivo results of stabilizing Aβ into non-toxic conformers and improving AD-related phenotypes in TgCRND8 transgenic mice [32,33], a cyclohexanehexol isomer, ELND005 (Scyllo-inositol), has been tested in a Phase 2 clinical trial [32]. Although the primary endpoints in the Phase 2 trial did not achieve statistical significance, ELND005 (250 mg, bid) demonstrated a biological effect on Aβ in CSF [34]. Currently, the sponsoring companies intend to advance this molecule into Phase 3 studies. To evaluate the biological effects of Aβo or Aβ aggregation inhibitors and to prove their mechanisms of action, a biofluid assay set-up would be a tool to find molecules and test their pharmacodynamic action. Previous research has reported that Aβo concentration measured by in-house ELISA method in CSF of AD patients is significantly higher than healthy controls [35]. Although there is criticism of the measurement of Aβo by single-antibody ELISA methodology, the effect of the anti-aggregant on Aβ oligomerization could be monitored by the measurement of Aβo in CSF. However, it is not clear that Aβo are detectable in CSF [36,37]. The recent development of tandem mass spectrometry (MS)-based quantitation of Aβ species in CSF may provide quantitative data for the effect not only of amyloidogenic secretases inhibitors but also Aβo inhibitors on APP metabolism in the brain [38].
Aβ clearance can be achieved by pumping Aβ out through the blood-brain-barrier (BBB) from the brain to the plasma and/or Aβ degradation by specific enzymes. There are two potential targets for enhancement of Aβ removal from the brain to the periphery: the receptor for advanced glycation end products (RAGE) and low-density lipoprotein receptor-related protein 1 (LRP-1) [39,40]. RAGE mediates the influx of Aβ into the brain, while LRP-1 mediates efflux of Aβ from the brain (Fig. 2). In addition to the possible role of RAGE-Aβ interaction for the activation of nuclear factor-κB signaling pathways, which may promote apoptosis and neuroinflammation [41], the RAGE-mediated influx of peripheral Aβ into the brain may increase amyloid load. A RAGE inhibitor or an LRP-1 activator may be a potential candidate for AD treatment based on amyloid clearance. In fact, an oral small molecule inhibiting RAGE activity (PF-04494700) has been tested in Phase 2 trials; however, the development was discontinued [42]. Another strategy to enhance Aβ clearance is the activation of Aβ degradation proteases, including neprilysin, insulin-degrading enzyme and plasmin [43,44]. However, specific activation of enzymes seems the more challenging approach than inhibition.
Immunotherapy against amyloid pathology
Two types of immunotherapy currently exist to enhance antibody-mediated Aβ clearance: active immunotherapy (anti-Aβ vaccine) and passive monoclonal anti-Aβ antibody treatment. The initial active immunotherapy with AN-1792 to induce anti-Aβ antibodies was not tolerable by the activation of cytotoxic T cells and autoimmune reaction followed by meningoencephalitis [45]. Subsequently, the more tolerable active immunotherapies using the improved design of an immunogen- containing the N-terminal, fragment of Aβ1-42 or N-terminus mimic peptide, including ACC-001, CAD106, V950, and Affitope AD02, are currently being tested in the early phases of clinical trials [46]. Passive immunotherapeutic approaches are also currently developing in parallel with active immunotherapy. To date, humanized monoclonal antibodies for passive immunotherapy are in clinical development. For example, an initial early phase clinical trial of bapineuzumab showed good tolerability with mild to moderate adverse events, including symptomatic and asymptomatic amyloid-related imaging abnormalities (ARIA) and improvement of exploratory efficacy measure (minimal mental status examination score; MMSE), when compared to the placebo group. However, the efficacy evaluated by prespecified primary endpoints (Alzheimer's Disease Assessment Scale-Cognitive; ADAS-Cog, or Disability Assessment for Dementia; DAD) in a Phase 2 trial did not show statistical significance, although a trend in favor of bapineuzumab was observed, particularly in apolipoprotein E4 noncarriers [47]. In addition, there were no observed treatment differences in either CSF Aβ or total tau, but the reduction of p-tau in the bapineuzumab group was significant when compared with the placebo group [48]. Although the clinical efficacy of bapineuzumab in the Phase 2 trial was not dramatic by week 52, amyloid removal from the brain was clearly observed in amyloid (PiB) PET scan [49]. In Phase 3 trial of bapineuzumab for 78 weeks, the clinical efficacy was disappointing although the changes of biomarkers were significant, particularly in ApoE carriers [50]. Another humanized monoclonal anti-Ab antibody, solanezumab (LY2062430) was applied to mild-to-moderate AD patients with an advantage of absence of significant ARIA [51]. Similar to bapineuzumab trial, however, solanezumab failed to have a success of clinical efficacy (phase 3 trial of EXPEDITION 1 and EXPEDITION 2 study) [52]. The disappointing results of intravenous bapineuzumab and solanezumab in phase III trial for mild to moderate AD patients would not necessarily exclude these from AD prevention trial, such as the Dominantly Inherited Alzheimer's Network (DIAN) study [53,54,55]. For passive immunotherapy using immunoglobulin, three small trials using intravenous immunoglobulin (IVIG) targeting multiple forms of Aβ suggest that IVIG can have favorable efficacy and will be tolerable [56,57,58]. However, two of these studies were not placebo-controlled studies, and another study was a retrospective case-control analysis. Recently, data of the clinical efficacy of IVIG in a placebo-controlled, multicenter Phase 2 clinical trial and the effect of IVIG on the concentration of plasma and CSF Aβ species have been published [59]. The results of this study showed an acceptable safety profile and a significant difference in plasma Aβ1-40 levels in the highest dose group when compared to the placebo group [60]. Recently, the disappointment of phase 3 clinical trial using IVIG in 390 patients with mild-to-moderate AD was announced, although a dose-dependent reduction in plasma Ab42, increase in plasma, CSF anti-oligomer and anti-fibril antibodies, and reduced brain fibrillar Ab42 was observed [61, and also see http://blog.alz.org/results-of-igiv-study-disappointing-but-not-discouraging].
Target related to tau hyperphosphorylation
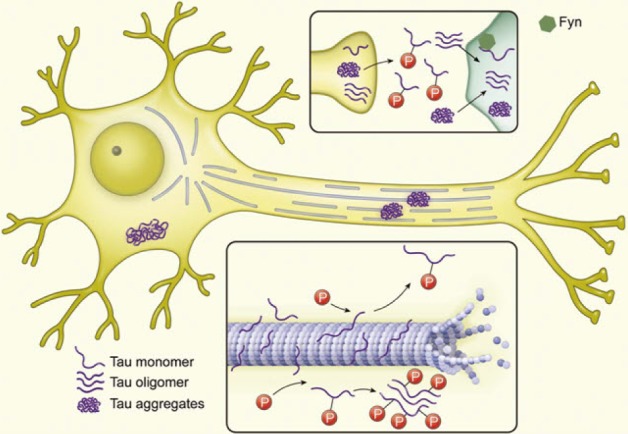
Intracellular fibrillary NFTs containing hyperphosphorylated and fibrillar species of tau are one of hallmarks of AD pathology, but they are not specific for AD. If tau is abnormally hyperphosphorylated and aggregated, it may be toxic due to gains of deleterious function or loss of the normal function of tau and microtubule instability followed by axonal transport failure (Fig. 3). There may be two approaches to inhibiting tau toxicity [11,62]. The first is the inhibition of abnormal hyperphosphorylation by targeting tau kinase and/or phosphatase. In addition, inhibitors of tau aggregation or disassemblers may be beneficial for protecting neurons from tau aggregate toxicity. There are several candidates of tau kinase and phosphatase related to tau hyperphosphorylation [63]. For example, glycogen synthase kinase 3 (GSK-3) is a well-known tau kinase, which is balanced with a phosphatase, protein phosphatase 2A. Valproic acid and lithium, well-known for the treatment of epilepsy and bipolar disorder, respectively, are GSK-3 inhibitors [64]. However, they didn't show consistent results for cognitive improvement or changes in CSF tau or p-tau181 concentration in clinical trials [65,66,67]. Several new GSK-3 inhibitors are being tested in clinical trials. NP031112 (tideglusib), which is a non-ATP competitive GSK3 inhibitor, reduces tau hyperphosphorylation and amyloid deposit, prevents neuronal death, and improves cognitive function in animal models [68]. This drug showed clinical benefits in a pilot, randomized, double-blind, placebo-controlled clinical trial, and it is currently being confirmed in a larger clinical trial [69]. Other multifunctional inhibitors of tau aggregation are also in development. Methylene blue, a famous histology dye, inhibits tau aggregation in addition to its antioxidant activity and the ability to enhance mitochondrial function. In a Phase 2, placebo-controlled study, a middle dose of methylene blue known as Rember improved cognitive function in mild-to-moderate AD patients and had evidence of slower progression of the disease [70]. Davunetide (an octapeptide) and nicotinamide are inhibitors of tau aggregation that showed prevention of cognitive deficit in AD animal models [71]. Rember is safe and tolerable and it continues in clinical studies but Davunetide failed in clinical trials for AD and other tauopathies (for a review, see http://www.alzforum.org/news/research-news/tau-targeting-drug-davunetide-washes-out-phase-3-trials).
CLINICAL PERFORMANCE OF CSF BIOMARKERS FOR EARLY DIAGNOSIS OF AD
By direct contact with extracellular space of the brain, CSF is the most useful biological fluid reflecting molecular events in the brain, which have driven intense research efforts to develop biochemical biomarkers for AD diagnosis in CSF. On the basis of prevailing scientific evidence and reliable clinical performance, CSF biomarkers have been involved in the recently published revision of AD diagnostic criteria for research purposes as supportive evidence for AD pathophysiology [3,4]. When we analyzed the overall clinical performance of CSF AD biomarkers (concentration of Aβ1-42, t-tau and p-tau181) for the diagnosis of AD in previously reported clinical studies, both mean sensitivity and specificity for Aβ1-42 and t-tau are over 80%, while those for p-tau181 are slightly lower than 80%. When Aβ1-42 and t-tau are combined (i.e., Aβ1-42/t-tau ratio or regression model using Aβ1-42 and t-tau concentration), both the mean sensitivity and specificity are higher than 85% [72]. Furthermore, longitudinal follow-up studies showed that CSF biomarker measurement has an ability to predict future development of AD in MCI patients [5,6,7,8,9,10]. The prediction of progression from MCI or preclinical AD to AD is important not only for the early diagnosis followed by therapeutic intervention but also for the design of clinical trials for developing disease-modifying therapies. In this context, large-scaled clinical studies are underway to test the clinical relevance of CSF biomarkers for early diagnosis of AD, even in the preclinical stage of AD development. For example, data from North American Alzheimer's Disease Neuroimaging Initiative (ADNI) showed the clinically reliable predictive performance of CSF AD biomarkers [8,73], using cut-off values determined by independent non-ADNI autopsy-confirmed samples [7]: the sensitivities of Aβ1-42 t-tau, p-tau181, and Aβ1-42/t-tau ratio are 96.4, 69.6, 67.9, and 85.7%, respectively, and the specificities are 76.9, 92.3, 73.1, and 84.6%, respectively,. North American ADNI is a multicenter, prospective, longitudinal, observation study for the evaluation of clinical characteristics, genetics, imaging biomarkers, and CSF AD biomarkers in healthy elderly subjects, MCI, and AD patients [74]. This study (ADNI-1) was completed in 2009, and ADNI-2 is ongoing. In ADNI-2, early amnestic MCI patients were added. The early amnestic MCI was defined as individuals meeting clinical criteria for amnestic MCI, who score between 0.5 and 1.5 standard deviation below the mean of normal healthy controls on delayed paragraph recall performance. Therefore, ADNI studies provided the long-term follow-up data for the changes in the initial diagnosis (e.g., early or late MCI to AD or normal to MCI). In addition to North American, European and Australian ADNI studies in western countries, Japanese, Chinese, and Korean ADNI studies are currently ongoing in Asia. Currently, numerous investigators are supporting the evidence that CSF biomarkers have a good diagnostic performance, particularly in combination with other biomarkers, including genetic biomarkers (e.g., ApoE genotype) and imaging biomarkers (e.g., hippocampal volume determined by MRI) [75,76,77,78]. However, there are several limitations for the application of CSF biomarkers for the diagnosis of AD in clinics distributed elsewhere. For example, the cut-off values for AD diagnosis are different across the studies due to interlaboratory variability in the measurement of CSF biomarker concentrations [73]. In other words, the causes of interlaboratory variability of CSF biomarker concentrations measured by immunoassay technologies were not yet completely elucidated (see below). To minimize the interlaboratory variability of CSF biomarker concentrations, global efforts, including the development of standardized protocols, reference materials and reference methods, are underway [79,80].
In addition, the amyloid hypothesis and chronological cascade relationships among amyloid and tau pathology, biomarkers of CSF level of Aβ1-42 and tau and imaging biomarkers, and clinical symptoms, still remain to be proven (Fig. 1), although no clear findings can negate this hypothesis. With the reliable clinical performance of CSF biomarkers, genetic and imaging biomarkers have a diagnostic value for early diagnosis of AD or prediction of disease progression. In particular, it is very important the elucidation of the neurodegenerative process and biomarkers reflecting the pathogenic process in preclinical stage of the disease, since it will accelerate the development of new therapeutics for AD treatment as well as the prevention of the disease [11,81]. To do this, the long-term longitudinal study observing the changes of clinical parameters, and CSF and imaging biomarkers in population with various degree of the disease will be required. Currently, ADNI-GO and ADNI-2 studies including early MCI subjects as well as normal healthy elderly subjects, MCI and early AD patients are underway.
CSF BIOMARKERS FOR AD TRIALS
The use of biomarkers in clinical trials depends on the mechanism of action of the developing therapy, goal of the trial, question to be solved, and stages of the trial. There are several purposes to include AD biomarkers in clinical trial design. First, they can be included in clinical trials as surrogate endpoints for the evaluation of the effects of a potential, new, disease-modifying therapy. Unlike cardiovascular or cancer clinical trials, in which the primary endpoint is typically the occurrence of a specific event, the use of mortality as an endpoint is not feasible for AD clinical trials due to the very large required sample size or follow- up period. If the measurement of biomarkers is valid, biomarker outcome measures can provide the evidence for the biochemical and/or molecular effects of a new therapy. In addition, biomarkers can be used as surrogate endpoints in which clinical measurements are not available, e.g., very early pre-symptomatic stage of AD. Although much work remains to be performed for the application of biomarkers as surrogate endpoints, the measurement of valid biomarkers has a potential to decrease the time that is required for evaluating clinical efficacy. To this end, it is necessary to determine whether the measurement of AD CSF or imaging biomarkers correlate with clinical endpoints and predict future clinical benefit or decline. In an AN1792 Aβ immunization Phase 1 trial, a small portion of antibody responders showed greater atrophy and progression of dementia despite the apparent reduction of amyloid load at autopsy [82,83]. However, it was evident in a larger Phase II trial that AN1792 immunization has a long-term functional benefit in antibody responders [84]. Among the current key unknowns are the time frames for affecting CSF biomarker measurement and what degree the biomarker change might relate to the clinical outcome. Therefore, we need to carefully consider the qualification and validation of AD biomarkers before using them as surrogate markers for clinical endpoints.
Second, biomarkers can be used diagnostically with clinical workup for inclusion and exclusion criteria. In particular, in early phase trials aiming to show mechanistic proof of concept, biomarker measurements can be used as a stratification tool for the pharmacodynamic evaluation of a drug. For example, the effects of a drug targeting brain amyloid reduction (e.g., anti-Aβ immunotherapy or secretase inhibitors) can be assessed in subjects with a high amyloid load who are differentiated by CSF or imaging biomarkers. In recent clinical trials, BMS-708163, a Notch-sparing γ-secretase, showed a good tolerance and a dose-dependent decrease of Aβ1-40 and Aβ1-42 in the CSF of healthy volunteers and AD patients [15,17,85]. As described above, the diagnostic performance of CSF biomarkers is highly reasonable for early diagnosis of AD and for the prediction of MCI progression, although further evidence should be accumulated for the diagnosis of preclinical AD. In fact, several recent disappointing results of clinical trials involving patients at the mild-to-moderate stage of AD emphasize the need for biomarkers that detect AD pathology in the early MCI stage or even at the pre-symptomatic stage. In the case of MCI, CSF AD biomarkers (e.g., diminished Aβ1-42 and elevated t-tau and p-tau181) can differentiate between those patients with underlying AD as the cause of the cognitive impairment from those MCI patients categorized in an early stage of non-AD dementia, patients with stable MCI for long periods of time, or patients in a group who will recover to normal cognition [86]. Disease-modifying drugs are likely to be most effective when they are given early in the pathogenic process of AD. Therefore, if patients in the early reversible stage of the disease can be differentiated from normal subjects by CSF and/or imaging biomarkers combined with clinical diagnosis, the possibility to have success in a clinical trial of disease-modifying therapy will increase.
Finally, biomarkers can be used for enrichment of study subjects, particularly in Phase 2 and 3 trials, and for increments of statistical power. In fact, AD patients with more extensive cortical atrophy, with low CSF Aβ1-42 level, or with genetic risk factors (e.g., ApoE4 allele) progress more rapidly than others [87]. A placebo-controlled trial including rapidly progressing subjects may enhance the opportunity to observe the drug-placebo difference within the follow-up time frame of the trial. In addition, the involvement of CSF and imaging biomarkers in clinical trial design may minimize the possible clinical variability in subjects who are recruited by clinical diagnosis alone. Furthermore, the statistical power of clinical trial design may increase by inclusion of biomarkers as baseline covariates to assess the treatment effects. As a consequence of low predictability of the progression to AD from MCI by clinical assessment alone, the inclusion of MCI patients in a treatment trial without the benefit of a biomarker-based assessment is likely to obscure a clinical outcome or require an increased sample size and longer observation. The enrichment and stratification strategy for recruitment of patients by inclusion of CSF AD biomarkers and/or other biomarkers will likely improve sample homogeneity and statistical power, and therefore, it would allow for a substantial reduction in sample size and cost-saving in clinical prevention trials or clinical trials of AD-modifying therapy in AD or MCI patients [69,88,89]. There are several prospective studies to evaluate the predictability of CSF AD biomarkers for AD progression from MCI with the realistic sample size and follow-up duration for what might be considered in Phase 2 or 3 clinical trials and with highly standardized protocols [5,7,90]. Despite the difference and lack of formal comparison, they agreed that CSF AD biomarkers showed a good diagnostic performance for MCI due to AD. Therefore, there is a trend to voluntarily include the CSF AD biomarkers in the design of current clinical trial for various purposes.
FUTURE PERSPECTIVES
Given the clinical performance of CSF AD biomarkers, many of the current AD clinical trials are focusing on the beneficial roles of biomarkers in the design of a cost-effective clinical trial in addition to the understanding of disease progression. Although recent results of a solanezumab trial argued the lack of correlation between CSF biomarker changes and treatment effect, biomarkers have the potential to be extremely useful for various purposes in clinical trials. We have learned from the massive amounts of data provided by ADNI and related projects around the world that there is still much to learn about how well CSF, imaging and genetic biomarkers reflect pathology and clinical manifestation. In addition, it might be important to determine the precision, reproducibility, and the causes of interlaboratory variability in the measurement of CSF AD biomarkers. For standardization, it will be important and consistent with the field to emphasize major standardization efforts like North American ADNI, started in 2004, and world-wide ADNI and other Alzheimer's Association-sponsored studies. The global collaborative efforts of investigators from academia, industry, and regulatory agencies to minimize the current analytical issues will likely position CSF biomarkers to become a contributing factor for successful clinical trials and development of new AD therapies.

 XML Download
XML Download