

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The prevalence of nonalcoholic fatty liver disease (NAFLD) has been reported to be approximately 20% in South Korea [1]. NAFLD is associated with insulin resistance and metabolic syndrome, such as obesity, type 2 diabetic mellitus (T2DM), and dyslipidemia [2]. NAFLD represents a broad spectrum ranging from simple steatosis to nonalcoholic hepatosteatosis (NASH), cirrhosis, and hepatocelluar carcinoma [3,4]. At the beginning stage of NAFLD, simple steatosis is a reversible condition of accumulation of lipid droplets in hepatocytes. Disorders in fatty acid metabolism appear to contribute to fat accumulation, such as increased influx of fat to the liver, increased de novo lipogenesis, decreased oxidation of fatty acids, and decreased secretion of very low-density lipoprotein [5,6]. Moreover, fat releases inflammatory cytokines and leptin, which promote lipolysis from adipose tissues and increase circulating free fatty acid (FFA) in plasma, causing dyslipidemia [7].
A high-fat diet and low physical activity induce visceral adiposity and obesity, and accumulation of fat inhibits the reaction of insulin in muscle and adipose tissue by decreasing the expression of glucose transporter 4 (GLUT4) and causes insulin resistance [8,9,10]. Additionally, insulin-dependent GLUT4 in the liver is also expressed in endothelial cells and hepatic stellate cells (HSCs) [11]. Leptin-induced HSC activation is associated with hepatic steatosis and fibrosis [12].
Exendin-4 (Ex-4) shows 53% amino acid homogeneity to mammalian glucagon-like peptide (GLP)-1, an incretin hormone, and shows agonist activity at GLP-1 receptors (GLP-1R) in vitro [13,14,15,16]. GLP-1, a hormone secreted by the L cells of the intestine, has numerous biological effects, including glucose-dependent enhancement of insulin secretion, suppression of glucagon secretion, delay of gastric emptying, improvement of satiety by acting on the hypothalamus, promotion of pancreatic β cell proliferation, and reduction of hepatic fat [17,18,19,20,21,22]. Exenatide is a synthetic analog of Ex-4 that does not function as a direct insulin sensitizer but reduces clinically significant body weight, possibly resulting in an insulin-sensitizing effect [23]. The expression of hepatic GLP-1R has been reported, and some studies suggest that GLP-1R agonists may play a role in hepatic metabolism [24]. GLP-1R agonists have been reported to exert effects on the liver, including an increase in lipolysis, a reduction in lipogenesis, and improvement in hepatic fibrosis [25,26,27]. However, the mechanism of the protective effects of Ex-4 on the reduction of hepatic steatosis is not well known.
Therefore, in the present study, we focused on the mechanism underlying the protective effects of Ex-4 on GLUT4 expression in the liver of leptin-resistant ob/ob mice.
Go to : 
METHODS
Animals
Male wild-type (WT) or ob/ob mice (4 weeks old) were purchased from Central Laboratory Animal Inc. (Seoul, South Korea) and maintained in the animal facility at Gyeongsang National University. The experiments were performed in accordance with the National Institutes of Health Guidelines on the Use of Laboratory Animals. The University Animal Care Committee for Animal Research of Gyeongsang National University approved the study protocol. Mice were individually housed using an alternating 12-h light/dark cycle. Mice, starting at 10 weeks of age, were randomly divided into four groups (n=10 mice per group). According to a previous study [22], WT or ob/ob mice were treated for 2 weeks with either Ex-4 (10 µg/kg; Byetta, Amylin Pharmaceuticals, Inc., CA, USA) every 12 h for the first 2 weeks. This treatment represented the induction phase. Respective control mice (WT and ob/ob) received saline every 12 h. After 2 weeks, Ex-4 treatment groups were treated with 20 µg/kg Ex-4 every 24 h for 8 weeks. Mice were weighed monthly and immediately prior to sacrifice at 20 weeks of age.
Glucose tolerance test (GTT) and insulin tolerance test (ITT)
Mice were fasted overnight (16 h) before the GTT. D-glucose (2 g/kg; Sigma-Aldrich, St. Louis, MO, USA) was injected intraperitoneally, and blood samples were taken before and 30, 60, 90, and 120 min after the injection of glucose. Blood glucose levels were measured using an Accu-Chek glucometer (Roche Diagnostics GmbH, Mannheim, Germany). The ITT was performed on mice around 2:00 PM. Mice were injected with insulin (0.75 U/kg; Humulin-R, Eli Lilly, Indianapolis, IN, USA) in 0.1 ml of 0.9% normal saline. A drop of blood was taken from the tail vein before and 15, 30, 45, and 60 min after the injection of insulin for the determination of blood glucose levels with a glucometer (Accu-Chek).
Measurement of serum metabolic parameters
For serum analysis, all mice were intramuscularly anesthetized with zoletil (5 mg/kg; Virbac Laboratories, Carros, France). Blood samples were extracted transcardially through the apex of the left ventricle with a 1-ml syringe and were allowed to clot for 2 h at room temperature. After centrifugation, the serum samples were removed and stored at -80℃ until analysis. Serum aspartate aminotransferase (AST), alanine aminotransferase (ALT), free fatty acid (FFA), total cholesterol, and triglyceride (TG), insulin, leptin, and adiponectin levels were determined using enzymatic colorimetric assays from Green Cross Reference Laboratory (Yongin-si, South Korea).
Liver TG colorimetric assay
After serum extraction, livers were stored at -80℃ until analysis. Liver TG concentrations (n=6 mice per group) were measured using the TG colorimetric assay kit (Cayman Chemical Company, Ann Arbor, MI, USA) according to the manufacturer's protocol.
Tissue collection and sample preparations
For tissue analysis, mice (n=4 per group) were anesthetized with zoletil (Virbac Laboratories) and then perfused transcardially with heparinized saline followed by 4% paraformaldehyde in 0.1 mL of phosphate-buffered saline (PBS). Six h after postfixation in the same fixative, livers were processed for paraffin embedding and sectioned (5 µm). Liver sections were stained with hematoxylin and eosin (H&E). The sections were visualized under a BX51 light microscope (Olympus, Tokyo, Japan), and digital images were captured and documented.
Oil red O staining
To determine hepatic steatosis, frozen liver sections (10 µm) were stained with 0.5% Oil red O (Sigma) for 10 min, washed, and counterstained with Mayer's hematoxylin (Sigma) for 30 s. The sections were visualized under a light microscope (Olympus), and digital images were captured and documented.
Western blot analysis
For protein extraction (n=6 per group), frozen liver tissues were homogenized in lysis buffer [15 mM HEPES (pH 7.9), 0.25 M sucrose, 60 mM KCl, 10 mM NaCl, 1 mM ethylene glycol tetraacetic acid, 1 mM phenylmethylsulfonyl fluoride, and 2 mM NaF]. Antibodies specific to the following targets were used: GLP-1R, CTGF, GLUT2, GLUT4, and PPAR-α from Abcam (MA, USA). The membranes were probed with each antibody or anti-β-actin (Sigma) and visualized using an enhanced chemiluminescence substrate (Pierce, Rockford, IL, USA). The Multi-Gauge v3.0 image analysis program (Fujifilm, Tokyo, Japan) was used to measure band density.
Immunohistochemistry
Deparaffinized liver sections were placed in a solution of 0.3% H2O2 for 10 min. After washing, sections were treated with diluted blocking serum for 20 min. Slides were incubated overnight at 4℃ in a humidified chamber with anti-rabbit GLUT4 (1:100; Abcam) diluted in blocking serum. After washing three times with 0.1 M PBS, sections were incubated for 1 h at room temperature with a secondary biotinylated antibody (1:200). The sections were washed and incubated in avidin-biotin-peroxidase complex solution (Vector Laboratories, Burlingame, CA, USA). Sections were then developed with 0.05% diaminobenzidine solution (Sigma) containing 0.05% H2O2, dehydrated through graded alcohols, cleared in xylene, and coverslipped with Permount (Sigma). Sections were visualized under a BX51 light microscope.
Statistics
Differences between WT and ob/ob mice with or without Ex-4 were determined by Student's t-test or one-way analysis of variance with Bonferroni post-hoc analysis. Values are expressed as the mean±standard error of the mean (SEM). p values less than 0.05 were considered as statistically significant.
Go to :
RESULTS
Effects of Ex-4 on the whole body, liver, and intraabdominal fat weight in ob/ob mice
After 6 weeks of Ex-4 injection, the body weight of ob/ob mice was reduced, whereas the same treatment did not affect the body weight of WT mice (Fig. 1A and B). To determine the effects of Ex-4 on liver and intraabdominal fat weight in ob/ob mice, we measured the weight of the liver, and perirenal and epididymal fat pads (Fig. 1C and D). Ex-4 significantly reduced the liver and intraabdominal fat weight in ob/ob mice (Fig. 1D). Particularly, the weight of perirenal fat around the kidney was significantly reduced by Ex-4.
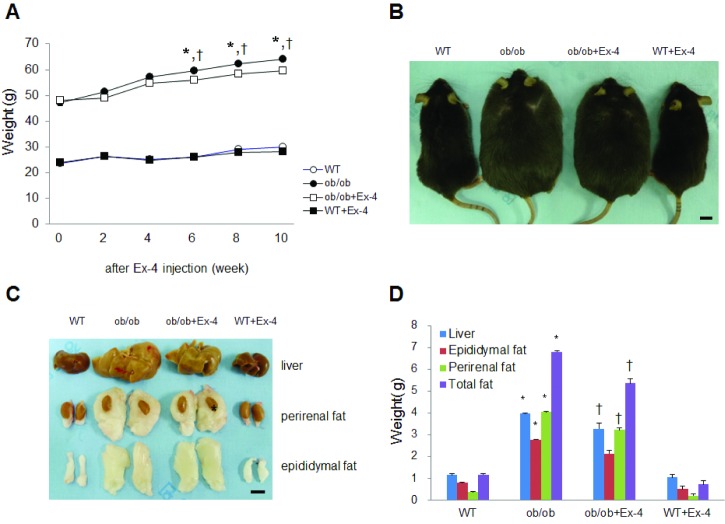 | Fig. 1Effect of long-term Ex-4 treatment on the weight of body, liver, and epididymal fat padsin ob/ob mice. (A) The change in body weight for 10 weeks after Ex-4 injection. (B) Representative photographs of WT, ob/ob, ob/ob+Ex-4, and WT+Ex-4 mice. Scale bar=1 cm. (C) Representative photographs of the liver, perirenal fat pads, and epididymal fat pads from each experimental group. Scale bar=0.5 cm. The asterisk indicates kidney within perirenal fat. (D) Graphs showing weights of the liver, perirenal fat pad, and epididymal fat pad, and total fat (perirenal+epididymal fat) from each experimental group. Data (n=10 mice per group) are presented as the mean±SEM. *p<0.05; vs. WT; †p<0.05 vs. ob/ob.
|
Effects of Ex-4 on serum adipokines and lipid parameters in ob/ob mice
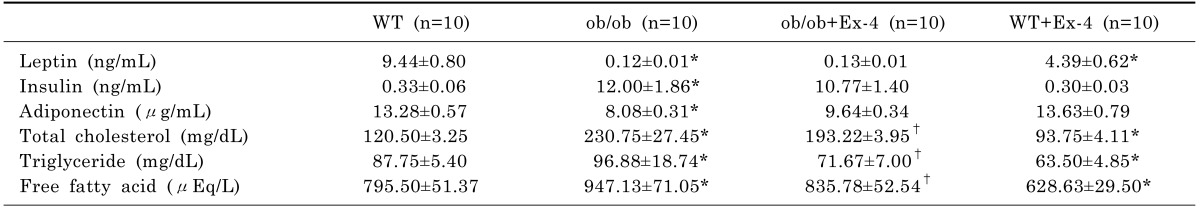
To determine the effects of Ex-4 on serum adipokines in ob/ob mice, we measured the concentration of insulin, adiponectin, and leptin using enzyme-linked immunosorbent assay (ELISA) (Table 1). The ob/ob mice with leptin deficiency showed low leptin levels compared with WT mice. However, Ex-4 did significantly decrease serum leptin levels in WT mice. There was also hypoadiponectinemia and hyperinsulinemia in ob/ob mice. However, these changed levels were not significantly reversed by Ex-4 treatment. As shown in Table 1, the serum total cholesterol, TG, and FFA levels in ob/ob mice were significantly reduced by Ex-4 treatment (p<0.05).
Effects of Ex-4 on insulin sensitivity in ob/ob mice
Next, to explore the role of Ex-4 in insulin resistance in ob/ob mice, the GTT and ITT were performed (Fig. 2). The insulin and glucose tolerance were significantly increased at 20-weeks-old ob/ob mice. However, administration of Ex-4 significantly improved insulin sensitivity in ob/ob mice (Fig. 2A) but not glucose tolerance (Fig. 2B).
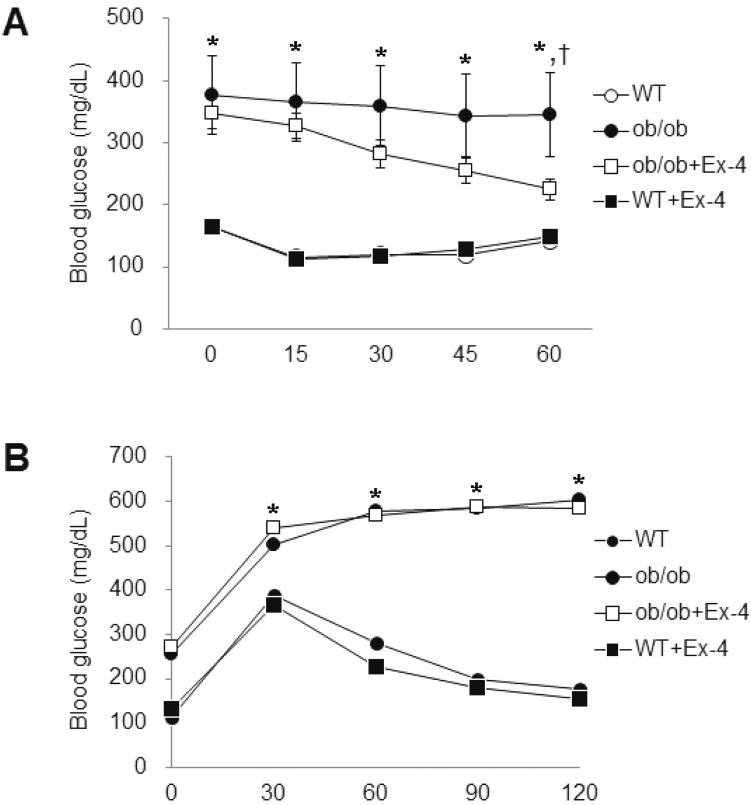 | Fig. 2Effect of Ex-4 on insulin and glucose tolerance in ob/ob mice. (A) Insulin glucose tolerance test. Blood glucose levels after insulin (0.75 U/kg) injection. (B) Glucose tolerance test. Blood glucose levels after D-glucose (2 g/kg) injection. Data (n=10 mice per group) are presented as the mean±SEM. *p<0.05; vs. WT; †p<0.05 vs. ob/ob.
|
Effect of Ex-4 on hepatic steatosis and fibrosis in ob/ob mice
To investigate whether Ex-4 affects hepatic steatosis and function in ob/ob mice, we performed H&E and Oil red O staining (Fig. 3A). As shown in Fig. 3A, the accumulation of fat and large distended lipid droplets appeared in the liver of ob/ob mice, which were remarkably reduced by Ex-4 treatment. To evaluate the effect of Ex-4 on liver function in the ob/ob mice, we measured serum AST and ALT levels (Fig. 3B). The effect of Ex-4 on hepatic TG levels in ob/ob mice was confirmed using a liver TG colorimetric assay (Fig. 3B). The serum levels of two hepatic enzymes and liver TGs were elevated in ob/ob mice compared with those of WT mice, whereas the levels were decreased in WT mice treated by Ex-4 (p<0.05).
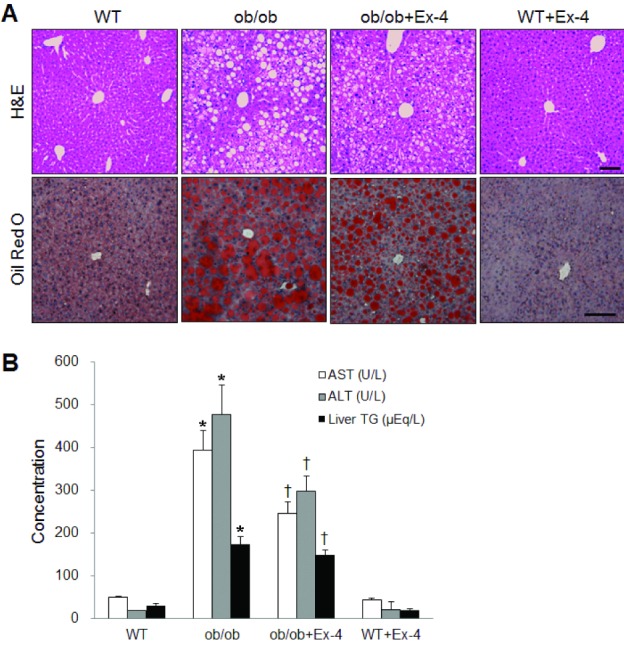 | Fig. 3Effect of Ex-4 on hepatic function and steatosis in ob/ob mice. (A) Representative microphotographs of H&E and Oil red O staining of liver sections from WT or ob/ob with or without Ex-4. Scale bar=100 µm. (B) Serum levels of AST, ALT, and liver TG from each experimental group. Data (n=10 mice per group) are presented as the mean±SEM. *p<0.05; vs. WT; †p<0.05 vs. ob/ob.
|
To examine the effects of Ex-4 on hepatic fibrosis in 20-week-old ob/ob mice, Masson trichrome staining was performed (Fig. 4A). A characteristic fibrosis pattern due to deposition of collagen along the sinusoids surrounding the hepatocytes was observed in ob/ob mice. Masson trichrome-stained collagen deposits were not observed in the liver of ob/ob mice treated with Ex-4. Similarly, the effect of Ex-4 on hepatic CTGF expression in ob/ob mice was confirmed by Western blot analysis (Fig. 4B). Hepatic CTGF expression levels were significantly higher in ob/ob mice than in WT mice (p<0.05), whereas the expression levels were significantly decreased CTGF expression in ob/ob mice treated by Ex-4 (p<0.05).
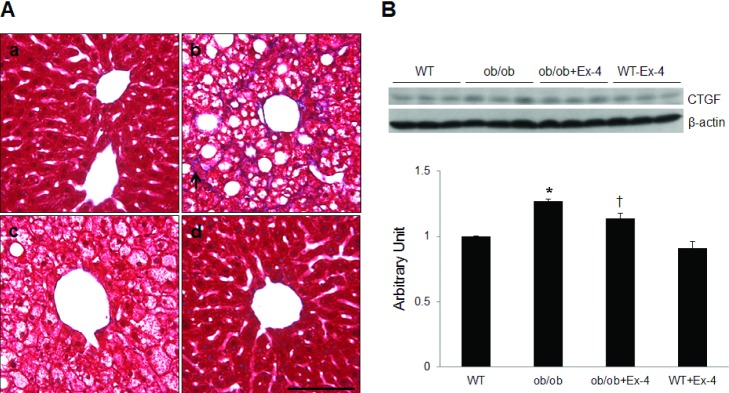 | Fig. 4Effect of Ex-4 on hepatic fibrosis in ob/ob mice. (A) Representative microphotographs of Masson trichrome staining of liver sections from WT (a), ob/ob (b), ob/ob+Ex-4 (c), and WT+Ex-4 (d) mice. Scale bar=100 µm. The asterisk indicates fibrosis. (B) Western blots showing hepatic CTGF expression and its quantification. Densitometry values for CTGF expression were normalized to β-actin expression and are represented as arbitrary units. Data are shown as the mean±SEM. *p<0.05; vs. WT; †p<0.05 vs. ob/ob.
|
Effects of Ex-4 on hepatic PPAR-α , GLP-1R, GLUT2, and GLUT4 expression in ob/ob mice
Next, we evaluated the effect of Ex-4 on hepatic PPAR-α expression in ob/ob mice (Fig. 5A). We found that PPAR-α expression in ob/ob mice was decreased compared with that in WT mice, whereas its expression was slightly increased by Ex-4 treatment. Similar to the previous report that Ex-4 plays as an agonist of GLP-1R, Ex-4 strongly attenuated and rescued the reduction of hepatic GLP-1R expression in ob/ob mice (Fig. 5B). Also, we showed the effect of Ex-4 on GLUT2, an abundant glucose transporter in the liver, in ob/ob mice. Western blot analysis showed that hepatic GLUT2 expression in ob/ob mice was increased compared with that in WT mice, whereas Ex-4 did not affect GLUT2 expression levels (Fig. 5C). Finally, to test whether Ex-4 affects GLUT4 expression in the liver of ob/ob mice, we performed Western blot analysis and immunohistochemistry (Fig. 5D and E). Hepatic GLUT4 expression was significantly lower in ob/ob mice than in WT mice, and GLUT4 expression was significantly increased by Ex-4 treatment. In accordance with the Western blot results, immunohistochemistry showed that GLUT4-positive cells were stained more intensely for GLUT4 in Ex-4-treated ob/ob mice than those in ob/ob mice (Fig. 5E).
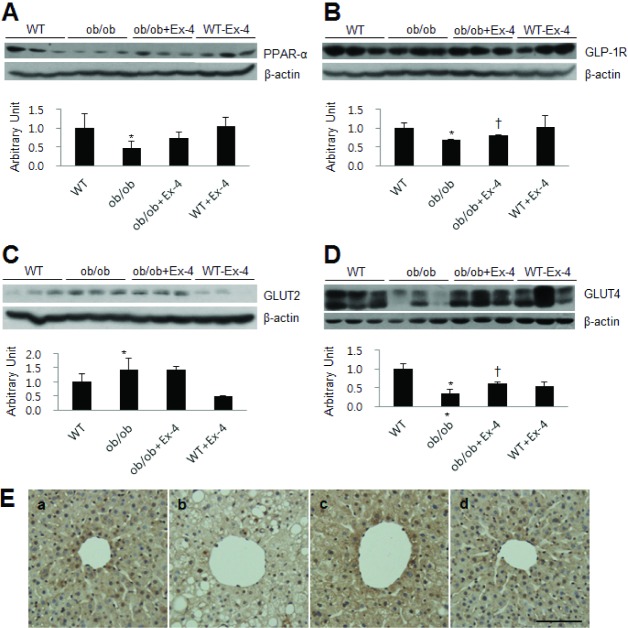 | Fig. 5Effect of Ex-4 on PPAR-α, GLP-1R, GLUT2, and GLUT4 expression in the liver of ob/ob mice. Western blots and quantifications showing hepatic PPAR-α (A), GLP-1R (B), GLUT2 (C), GLUT4 (D) expression. Densitometry values for each protein were normalized to β-actin expression and are represented as arbitrary units. Data are shown as the mean±SEM. *p<0.05 vs. WT; †p<0.05 vs. ob/ob. (E) Representative microphotographs of immunostained GLUT4 in liver sections from WT (a), ob/ob (b), ob/ob+Ex-4 (c), and WT+Ex-4 (d) mice. Scale bar=100 µm.
|
Go to :
DISCUSSION
The present study revealed that Ex-4 treatment reduced body and liver weight and improved hepatic steatosis and fibrosis in ob/ob mice. We demonstrated that long-term injection of Ex-4 reduced hepatic fibrosis regulating GLUT4 and CTGF and then decreased the rising NAFLD in ob/ob mice. The present results are coincided with previous studies, which reported a reduction in body weight by Ex-4 treatment [28]. At the age of 20 weeks, ob/ob mice showed an increase in visceral adiposity as well as body and liver weight. However, these increased parameters in ob/ob mice were significantly decreased by Ex-4 treatment for 10 weeks. The accumulation of visceral fat is a risk factor for insulin resistance, hepatic steatosis, and fibrosis [29]. We also showed that the levels of which are lipid metabolism parameters were rescued by Ex-4 treatment in ob/ob mice. Because ob/ob mice are congenitally deficient in leptin, Ex-4 had no effect on the serum leptin level in ob/ob mice but reduced the serum leptin level in WT mice. Importantly, Ex-4 reduced hepatic steatosis and fibrosis in ob/ob mice and improved hepatic functions.
Many studies have reported the effects of Ex-4 on hepatic steatosis [22,28], but the exact regulatory mechanisms involved in the protective effects of Ex-4 against hepatic steatosis or fibrosis in ob/ob mice remained elusive. Thus, in the present study, we showed GLP-1R as a target of Ex-4 to prevent or ameliorate the hepatic steatosis or fibrosis in ob/ob mice.
Recently, weight loss could lead to a reduction in hepatic steatosis and an increase of insulin sensitivity and affect directly GLP-1R in hepatocytes [30]. In patients with NASH, GLP-1R expression was decreased in the liver, and high fat diet-fed mice also showed a decrease in GLP-1R expression in the liver [28]. In the present study, reduced expression of GLP-1R in ob/ob mice was considered to be associated with metabolic syndrome such as obesity, hyperglycemia, and dyslipidemia. These data indicate that Ex-4 acts as an agonist of GLP-1R and increases GLP-1R expression in hepatocytes in leptin-deficient ob/ob mice.
β-oxidation of fatty acid is the process by which FFA is broken down to generate acetyl-coA, and is closely associated with PPAR-α function [31]. PPAR-α is a transcription factor and a major regulator of lipid metabolism in the liver. In ob/ob mice, the reduced expression of PPAR-α indicates the decreased β-oxidation in hepatocytes, along with reduction in FFA being broken down to acetyl-coA. Increased FFA combined with glycerol-3-phosphate in hyperglycemia results in the accumulation of TG droplets in hepatocytes [32]. Ex-4 enhances the expression of PPAR-α, promotes β-oxidation, and decreases TG in hepatocytes. We suggest that the activation of GLP-1R by Ex-4 may play an important role in increasing PPAR-α expression and β-oxidation [30]. Our data also showed that Ex-4 rescued the reduction of PPAR-α expression in ob/ob mice, and inhibited hepatic steatosis.
GLUTs are a group of membrane proteins that facilitate the transport of glucose across cellular membranes. Many isoforms of GLUTs (GLUT 1-6, 8-12) are expressed in the human liver [10]; among them, GLUT2 and GLUT4 are the major GLUTs responsible for glucose transport into hepatocytes [33]. The major role of hepatic GLUT2 is to regulate efflux rather than influx of glucose. Obesity-induced insulin resistance drives an increase in GLUT2 levels that may further exacerbate metabolic dysfunction in NALFD [34]. Our data showed that hepatic GLUT2 expression was increased in ob/ob mice compared with WT mice (Fig. 5B). GLUT4 is a major insulin-dependent glucose transporter and plays a rate-limiting role in glucose utilization in insulin-sensitive tissues, including brown and white adipose tissues and skeletal and cardiac muscles [33]. The present study showed that hepatic GLUT4 expression was lower in ob/ob mice than in WT mice, and the GLUT4 expression was increased by Ex-4 treatment (Fig. 5D). It has been demonstrated that Ex-4 can amplify insulin signaling in 3T3-L1 adipocytes by up-regulation of GLUT4 [35]. GLP-1R is a direct target of Ex-4 and leads to an increase in insulin sensitivity and stimulation of glucose uptake into hepatocytes by increased GLUT4 expression. This data indicate that Ex-4 may indirectly inhibit hepatic steatosis by regulating hepatic glucose metabolism. Thus, further study needs to investigate the direct molecular players of Ex-4 on GLUT4 regulation in hepatocytes. In our previous study, treatment of α-lipoic acid treatment improved insulin sensitivity and suppressed the inflammatory response in obese type 2 diabetic rats [36]. That study also showed that alpha-lipoic acid treatment increased hepatic GLUT4 expression and ameliorated hepatic steatosis [36]. We assumed that enhanced hepatic GLUT4 expression plays an important role in improving insulin sensitivity and suppressing the progression of hepatic steatosis.
GLUT4 is also expressed in endothelial cells and HSCs in the liver [10]. HSCs are known to be a primary source of extracellular matrix in collagen scar tissues, which are produced in response to liver injury [37,38]. Increased expression of CTGF is associated with HSC activation and progression [39]. Hyperglycemia and hyperinsulinemia are main factors in the progression of fibrosis in patients with hepatic steatohepatitis through the upregulation of CTGF [40]. At the age of 20 weeks in ob/ob mice, we found that mild hepatic fibrosis and increased CTGF expression compared with that in WT mice (Fig. 4). However, Ex-4 inhibited hepatic fibrosis and CTGF as well as hepatic steatosis. These data indicate that Ex-4 has an anti-fibrogenic effect in activated HSCs in ob/ob mice.
In conclusion, Ex-4 treatment improved hepatic steatosis by increasing GLP-1R and GLUT4 expression and inhibited hepatic fibrosis by decreasing CTGF expression in HSCs in ob/ob mice. Our data suggest that GLP-1 agonists are potential therapeutics for the treatment of obesity-associated hepatic steatosis and fibrosis by regulating GLUT4.
Go to :
 XML Download
XML Download