

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Dental pulp inflammation (pulpitis) is caused mainly by bacterial infections of the dentin or root canal [1]. In addition to caries-associated bacteria, pulp exposure to a chemical stimulus, mechanical stimulus, and trauma can trigger an inflammatory response [2]. Inflammation is a multifaceted response mediated by the activation of cells of the immune system. Cells of the immune system produce nitric oxide (NO) during inflammatory processes [3]. NO is a short lived, highly reactive free radical gas synthesized by nitric oxide synthase (NOS) through conversion of the guanidinic group of L-arginine to citrulline [4,5]. NO is a gaseous signaling molecule that regulates various physiological and pathophysiological processes in the human body. These processes include circulation and blood pressure, platelet function, host defense, and neurotransmission in the central nervous system and in peripheral nerves. It possesses cytotoxic properties that are aimed against pathogenic microbes, but it can also have damaging effects on host tissue [6].
Three different NOS isoforms have been characterized. The neuronal NOS (nNOS, NOS I) is expressed in the neurons of the brain and peripheral nervous system [7]. Endothelial NOS (eNOS, NOS III) is mainly expressed in endothelial cells [8,9]. Both nNOS and eNOS are constitutively expressed and are inactive in resting cells. The third isoform of the NOS family is the inducible NOS (iNOS, NOSII). No iNOS expression is found in most resting cells. Exposure to microbial products, such as lipopolysaccharide (LPS) and dsRNA or proinflammatory cytokines such as interleukin-1 (IL-1), tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) induces the expression of iNOS gene in various inflammatory and tissue cells [6]. These NOS isoforms also exist in the dental pulp tissue. nNOS is localized within the nerve fibers in the dental pulp, and NO produced by nNOS may act as a neurotransmitter within the pulp tissue [10]. eNOS is localized in endothelial cells and odontoblasts of the human dental pulp cells [11], indicating that NO synthesized by eNOS is responsible for regulation of the microcirculation within the pulp chamber and may control proliferation and differentiation of odontoblasts [12]. iNOS is expressed in the inflamed dental pulp cells and is mainly involved in inflammatory process and immune reaction [13-15]. The dental pulp cells abundantly express three NOS isoforms such as eNOS, nNOS and iNOS, which are known to play a pivotal role in physiologic and pathologic processes [11-13].
Apoptosis is an energy-dependent programmed cell death that is crucial to the balance between cellular survey and death. Previous studies have shown that mitochondrial dysfunction leads to an increase in expression of pro-apoptotic members of the B cell lymphoma 2 (Bcl-2) protein family as well as causes a loss of the mitochondrial membrane potential (MMP) and an increase in the release of cytochrome c from the mitochondrion to the cytoplasm. This process ultimately results in the activation of caspase-3, -6, and -7, as well as the activation of caspase-8, which cleaves Bid into tBid, which causes the release of apoptogenic proteins from the mitochondria and thus induces cell apoptosis [16-19].
In previous studies, NO has been shown to suppress apoptosis in endothelial cells [20], hepatocytes [21], eosinophils [22], and splenocytes [23], while inducing apoptosis in other cell types such as VSMCs [24], macrophages [25], neuronal cells [26], and pancreatic islet cells [27]. Large amounts of NO produced by NOS can induce apoptotic and necrotic cell death because NO can be cytotoxic at high concentrations in various cells including neuronal cells [28,29]. The molecular mechanism of the bifunctional action of NO remaines unclear and in controversy.
In general, NO acts as an intra- and intercellular messenger with various functions at the physiological level, whereas it can be cytotoxic at high concentrations, resulting in necrotic and apoptotic cell death [28,29]. Therefore, large amounts of NO synthesized by NOS can be cytotoxic to the dental pulp cells since previous studies have demonstrated that the inflamed pulp cells exhibit remarkably enhanced expression of iNOS which can produce large amounts of NO [14,15]. Nevertheless, NO-induced cytotoxicity in dental pulp cells and its underlying mechanism have not yet been elucidated. On the basis that the dental pulp cells abundantly express NOS, the present study aimed to investigate the mechanisms underlying NO-induced cell death of the HDPCs.
METHODS
Ethics statement
The study was approved by the Ethics Review Board of Chonnam National University. All the studies involving human participants were conducted in full compliance with government policies and the Declaration of Helsinki. All participants completed an informed consent.
Cell culture
HDPCs were obtained from the teeth of dental patients in Chonnam National Hospital. Teeth were immediately placed in phosphate-buffered saline (PBS) supplemented with antibiotics (100 µM/ml penicillin and 100 µg/ml streptomycin) and 0.25 µg/ml fungizone. The teeth were then transported to the laboratory on ice within 15 min of extraction. The teeth were sectioned horizontally at 1 mm below the cementoenamel junction (CEJ) using a #330 carbide bur mounted on a high-speed handpiece with an air-water spray and then they were split open. The pulp tissues were removed aseptically and minced with a blade into small fragments. They were then placed in a 6-well cell culture plate and incubated in DMEM (Gibco-BRL, USA) supplemented with 10% fetal bovine serum (FBS, Gibco-BRL, USA) and antibiotics. The cultures were maintained at 37℃ in a humidified atmosphere of 5% CO2. Cell cultures between the fifth and sixth passage were used in this study.
Cell viability assay
The effect of sodium nitroprusside (SNP, Sigma, USA) treatment on the cytotoxicity of HDPCs was determined by MTS assay. Breifly, cells were cultured overnight in 96-well plates (~1×104 cells/well). Cell viability was assessed after the addition of SNP at the indicated concentrations for 3, 6, 12 or 24 hrs in 48-well plates. The number of viable cells was assessed by determination of the A490 nm of the dissolved formazan product after addition of MTS for 1 hr at 37℃ as described by the manufacturer (Promega, USA).
Nuclear staining with 4'6'-diamidino-2-phenylindole (DAPI)
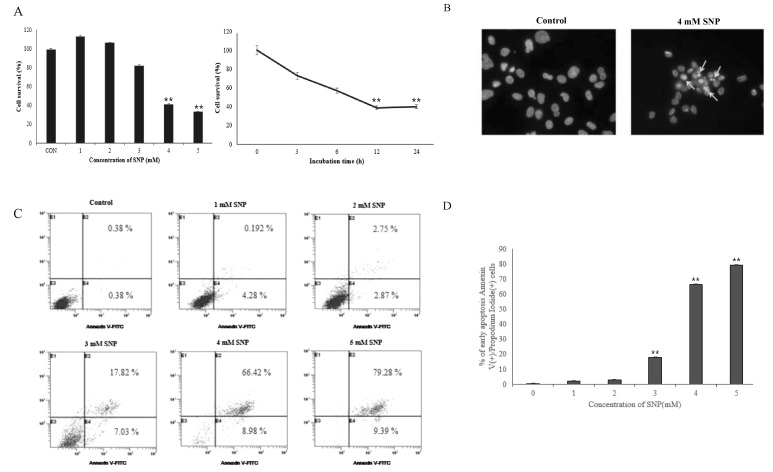
Morphological changes in apoptotic cells were investigated by 4'6'-diamidino-2-phenylindole (DAPI) staining. Cells were plated in an 8-well chamber slide at a density of 1×104 cells/ml and incubated for 24 hrs, subsequently followed by treatment with 4 mM SNP for 5 hrs. The cells were then washed with 1×PBS and fixed with 4% paraformaldehyde. After incubation for 5 min at room temperature (RT), cells were stained with 10 µg/ml of DAPI (Sigma, USA) in PBS and observed under an LSM 510 confocal microscope (Zeiss, Germany).
Annexin V cell assay
Cells undergoing apoptotiis was determined by Annexin V-FITC Apoptosis Kit II (Pharmingen, USA). Cells were treated with SNP (1, 2, 3, 4 and 5 mM) and incubated for 24 hrs, harvested at a density of 106 cells/ml and resuspended in 1×binding buffer. In a 5 ml tube, Annexin V-FITC and PI were added and incubated for 15 min at RT in the dark. Then, 1×binding buffer was mixed to each tube and stained cells were measured by flowcytometry (Beckman, Germany).
Detection of ROS production
Reactive oxygen species (ROS) production was monitored by a fluorescence spectrometer (Hitachi F-4500, Japan) using 2',7'-dichlorofluor-bescin diacetate (DCF-DA, Sigma, USA). Cells (5×104 cells/well) were plated in a 96-well plate for 24 hrs and incubated with 5 mM N-acetyl-cysteine (NAC, Sigma, USA) and SNP (1, 2, 3, 4 and 5 mM) treatment. After DCF-DA (25 µM) was treated and incubated for 15 min at 37℃, the fluorescence excitation was analyzed at 480 nm and the fluorescence emission was analyzed at 530 nm using a fluorescence spectrometer (Hitachi F-4500, Japan).
Assay for caspase activation
Caspase activities were determined by the caspase-3 and -9 activity assay kits (MBL, USA) according to the manufacturer's protocol. Briefly, HDPCs were grown on 100 mm dishes and treated with SNP for the indicated times (3, 6, 9, 12 and 24 hrs). The media were removed from the culture dishes and the cells were collected and washed with PBS, then resuspended in cell lysis buffer. After incubation on ice for 10 min, the lysates were centrifuged for 20 min at 11,000×g, and the supernatants were collected and protein concentrations were determined (BCA assay, Pierce). Fifty µl/ml of cell lysates were mixed with the reaction buffer containing the DEVD-pNA substrate (200 µM) for caspase-3 activity and the LEHD-pNA substrate (200 µM) for caspase-9 activity. After incubation for 24 hrs at 37℃, absorbance was measured in the wells at 405 nm by an ELISA reader.
Western blotting
Cells were washed twice with PBS and proteins were solubilized in the lysis buffer (500 mM Tris-HCl, pH 7.4, 150 mM NaCl, 5 mM EDTA, 1 mM Benzamide, 1 µg/ml Trypsin inhibitor) containing a protease inhibitor cocktail (Complete, Germany). Lysates were incubated for 30 min at 4℃, centrifuged at 11,000×g for 20 min at 4℃ and protein concentrations were determined by BCA protein assay (Pierce, IL). Protein extracts (50~100 µg) were boiled for 5 min in SDS-sample buffer and then subjected to electrophoresis on 12% polyacrylamide gel. Proteins were electroblotted onto a nitrocellulose membrane (Amersham Pharmacia Biotech, UK) and blocked with 5% skim milk (Becton Dickinson, USA) in tris-buffered saline-0.1%Tween 20 (TBS-T). Primary antibodies of rat monoclonal anti-cytochrome c (BD, USA), Bcl-2 (Santa Cruz, USA), Bax, cleaved caspase-3 (Cell signaling, USA) were used. After rinsing membrane three times in TBS-T and subsequently incubated with specific peroxidase-conjugated secondary antibodies (Sigma, USA) for 1 hr. Bound antibodies were visualized using an enhanced chemiluminescence detection system (Amersham Pharmacia Biotech, UK).
Statistical analysis
Each experiment was repeated at least three times unless stated otherwise. The data were expressed as means±S.D. The statistical significance of was performed using one-way and two-way analysis of variance (ANOVA) followed by a Student-Newman-Keuls test. Results were considered statistically significant when the p value was less than 0.05.
RESULTS
NO induces apoptosis in HDPCs
SNP, is widely used NO donor, that in vivo and in vitro releases NO [4]. To investigate the cytotoxic effect of SNP on HDPCs, cells were incubated with different concentrations of SNP (1, 2, 3, 4 and 5 mM) or 4 mM SNP for the indicated times (3, 6, 12 and 24 hrs). As shown in Fig. 1A, SNP was gradually attenuated cell viability of HDPCs in a dose- and time-dependent manner. The cell survival was less than 60% when the cells were treated with 4 mM SNP for 24 hrs. After exposure to 4 mM SNP for 5 hrs in HDPCs, cells showed typical apoptotic morphology including cell shrinkage, chromatin condensation and nuclear fragmentation (Fig. 1B). Annexin V is phosphatidylserine, which appears on the cell surface as a general indicator of apoptosis. SNP-treated HDPCs caused an increase in the number of Annexin V-positive cells in a dose-dependent manner (Fig. 1C). Together, these results strongly suggest that NO induces apoptosis in HDPCs.
NO-induced apoptosis in HDPCs is mediated by mitochondria-dependent pathway
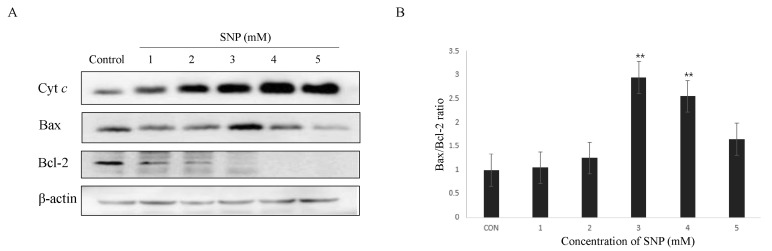
To validate whether mitochondria are involved in NO-induced apoptotic cell death of HDPCs, cytosolic cytochrome c level was assessed as a consequence of cytochrome c released from the mitochondria into cytoplasm. Cytosolic cytochrome c level was dose-dependently increased in response to exposure to SNP in HDPCs (Fig. 2A). Because NO increased the release of cytochrome c into the cytosol from the mitochondria in HDPCs, we determined if NO alters the level of Bcl-2 family, which are important regulators of mitochondrial permeability. The expression level of Bax, a pro-apoptotic member of the Bcl-2 family, was upregulated, whereas the expression of Bcl-2, an anti-apoptotic member of the Bcl-2 family, was downregulated in response to SNP treatment in HDPCs. Also, Bax/Bcl-2 ratio was increased in SNP-treated HDPCs compared to control (Fig. 2). These results demonstrate that NO-induced apoptosis is likely mediated by mitochondrial pathway in HDPCs.
NO-induced apoptosis in HDPCs is mediated by caspase-dependent pathway
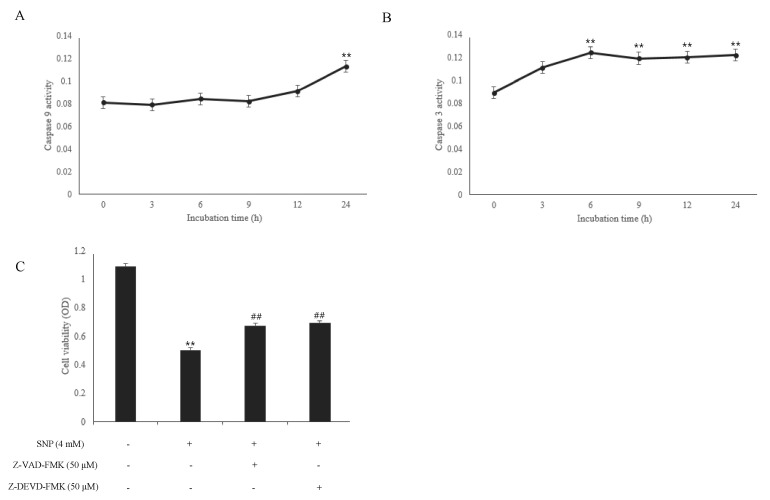
The release of cytochrome c from the mitochondria into the cytosol can activate caspases during apoptosis. SNP-treated HDPCs enhanced the activities of caspase-9, an initiator caspase, and caspase-3, an executioner caspase, in a time-dependent manner (Fig. 3A and B). In addition, to assess the role of caspases in NO-induced apoptosis, the specific inhibitor for caspase-3 (z-DEVD-fmk) and pan-caspase inhibitor (z-VAD-fmk) were pretreated for 30min before SNP treatment and MTS was performed. The results of the MTS assay showed that these caspase inhibitors significantly attenuated NO-induced cell death, indicating that these caspases play a crucial role in NO-induced apoptosis in HDPCs (Fig. 3C).
ROS is a mediator of NO-induced apoptosis in HDPCs
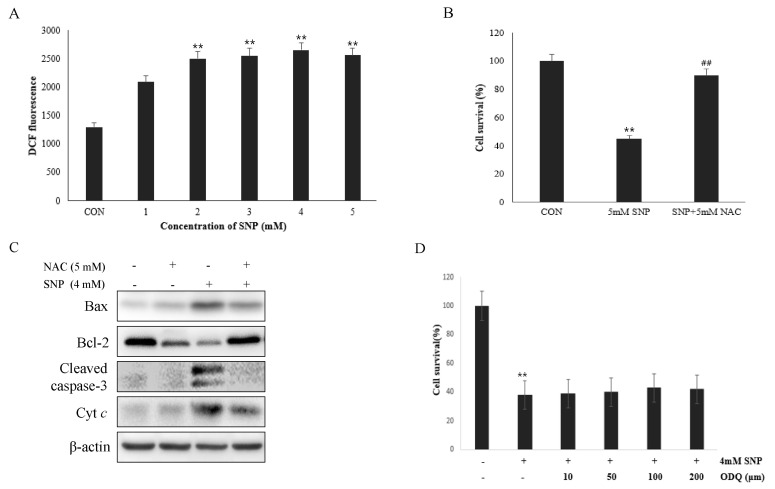
To determine the involvement of ROS in NO-induced cell death of the HDPCs, ROS production was measured using DCF-DA. SNP-treated HDPCs exhibited enhanced ROSproduction in a dose-dependent manner (Fig. 4A), whereas pretreatment of cells with 5 mM NAC, a ROS scavenger, alleviated the decrease in cell viability induced by SNP (Fig. 4B). These data suggest that ROS produced by NO plays as a crucial mediator in NO-induced apoptosis of HDPCs. To confirm the involvement of ROS in NO-induced apoptosis in HDPCs, cells were treated with 4 mM SNP alone or in the presence with 5 mM NAC. The expression levels of Bax, Bcl-2, cytochrome c and cleaved caspase-3 induced by SNP treatment were ameliorated by NAC (Fig. 4C).
In addition, NO can affect a variety of physiological processes, including programmed cell death (PCD) through cyclic guanosine monophosphate (cGMP)-dependent and -independent pathways. To examine whether NO acts through the cGMP pathway, effects of the soluble guanylate cyclase inhibitor on NO-induced cell death in HDPCs were examined. ODQ (100 µM), a guanylate cyclase inhibitor, did not rescue the cell viability decreased by SNP (Fig. 4D), indicating that NO-induced apoptosis was not mediated through the cGMP pathway.
DISCUSSION
Irreversible pulpitis has been associated with pain and an increase in the number of pulp inflammatory cells. Based on the action of NO elsewhere, NO may possibly participate in the sensory and autonomic innervation of the dental pulp, and may influence local inflammatory responses [14]. Indeed, the inflamed dental pulp cells exhibit high expression of iNOS which can result in mass production of NO [13-15]. However, the cellular mechanisms underlying NO-induced cell death in dental pulp cells was poorly understood.
In the present study, we investigated the effects of NO on growth in HDPCs. SNP, an NO donor, decreased the cell viability and caused apoptotic morphological changes including chromatin condensation, DNA fragmentation, and cell shrinkage in HDPCs. Besides, SNP treatment of HDPCs caused an increase in the number of Annexin V-positive cells in a dose-dependent manner. These results demonstrate that NO induces apoptosis in HDPCs.
Mitochondria-dependent apoptosis, which is upstream of caspase activation, is regulated by members of the Bcl-2 family. Bcl-2 family proteins regulate the release of apoptogenic cytochrome c by the mitochondrial channel VDAC [30,31]. The Bcl-2 subfamily contains anti-apoptotic proteins such as Bcl-2 and Bcl-XL, which reduce cytochrome c release and loss of mitochondrial transmembrane potential (Δψm), whereas the Bax subfamily contains pro-apoptotic proteins such as Bax and Bak, which induce cytochrome c release and loss of Δψm [32-34]. Therefore, the ratio of pro-apoptotic to anti-apoptotic protein may be pivotal for the release of cytochrome c from mitochondria into the cytosol. In the present study, expression of Bax was increased and level of Bcl-2 was decreased in HDPCs, indicating that increased ratio of Bax to Bcl-2 in NO-induced apoptosis of HDPCs mediates the transition of cytochrome c from the mitochondria into the cytosol. These results suggest that NO alter the expression of the Bcl-2 family in HDPCs, which affects the permeability of the mitochondrial membrane and the release of cytochrome c from the mitochondria to the cytosol. These findings definitely demonstrate that NO-induced apoptosis in HDPCs is mediated by the mitochondria-dependent pathway. The present findings along with those from previous studies suggest that the change in the Bax/Bcl-2 ratio is an important factor of NO-induced apoptosis. However, the exact mechanism of how NO induces a change in the Bax/Bcl-2 gene expression is unclear. Some investigators have suggested that p53 is a mediator between NO and Bax/Bcl-2 genes because NO can induces the accumulation of p53 [35,36], which activates a direct transcription of the Bax gene and inhibits a transcription of the Bcl-2 gene in macrophages [37,38]. However, this may not be true for HDPCs and this issue needs to be investigated in further study.
A variety of free radicals such as ROS and peroxynitrite are known to impair mitochondrial function, subsequently resulting in loss of mitochondrial transmembrane potential and release of mitochondrial pro-apoptotic molecules including cytochrome c, Smac, apoptosis-inducing factor (AIF) [39,40]. The present study showed that SNP treatment resulted in an increase in cytochrome c release from the mitochondria into the cytoplasm in a dose-dependent manner. The released the cytoplasmic cytochrome c was reported to be an upstream mediator of caspase-3 activation in different cell types [30,41,42]. In general, caspase-3 is one of the key, common proteases in the mitochondria-dependent pathway and it is particularly important in free radical-induced apoptosis [43-45]. The present study showed that caspase-9 and -3 activities were enhanced in SNP-treated HDPCs. Additionally, pretreatment with z-DEVD-fmk (caspase-3 inhibitor) and z-VAD-fmk (pan-caspases inhibitor) significantly inhibited NO-induced apoptosis, indicating that NO-induced apoptosis in HDPCs is mediated by caspase-dependent pathway. However, there are some previous reports that a caspase-independent pathway such as the MAP kinase pathway has also been proposed to be involved in NO-induced cell death in HL-60 cells and PC12 cells [46,47].
Previous studies have shown that NO causes apoptosis through the cyclic GMP pathway [48] or can induce the production of ROS such as H2O2 in mitochondria and its reaction with superoxide, resulting in formation of peroxynitrite in various other cells [46,49]. In the present study, SNP treatment increased the production of ROS, whereas NAC, a free radical scavenger, rescued the cell viability of HDPCs decreased by SNP. Besides, SNP-induced activation of proapoptotic mitocondria-dependent pathways (Cytochrome c, Bax, Bcl-2, and caspase-3) is down-regulated in the presence of NAC, a ROS scavenger. Meanwhile, ODQ did not rescue the cell viability of HDPCs decreased by SNP. From these results, it can be speculated that NO-induced apoptosis in HDPCs may be mediated in part by ROS, but not by cyclic GMP.
In conclusion, the present study shows that NO induces apoptosis in HDPCs through the mitochondria-dependent pathway mediated by ROS and Bcl-2 family, but not by the cyclic GMP pathway.
 XML Download
XML Download