

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Mast cells play a central role in the innate immune response. They are particularly important in helping to recruit other cells, such as basophils, neutrophils and lymphocytes. Mast cells play an important role in a number of allergic diseases including atopic dermatitis and asthma, and contribute to chronic inflammatory conditions like atherosclerosis, vasculitis and rheumatoid arthritis. Mast cells have a number of secretory granules including histamine and 5-hydroxytryptamine, and rapidly synthesize lipid-derived mediators, such as prostaglandins and leukotrienes from arachidonic acid (C20:4: ω-6) released by the activation of phospholipase A2 [1]. The addition of arachidonic acid to RBL-2H3 cells distinctly alter the fatty acid composition of the cell membrane and enhance Fcε RI-mediated degranulation and tumor necrosis factor-alpha (TNF-α) release with augmented tyrosine phosphorylation of some proteins and Ca2+ influx [2]. We previously reported that exogenous arachidonic acid significantly increases histamine release from RBL-2H3 cells [3].
On the other hand, ω-3 polyunsaturated fatty acids (PUFAs) reportedly decrease the production of arachidonic acid-derived eicosanoids. These fatty acids decrease the production of inflammatory cytokines, such as TNF, interleukin (IL)-1, and IL-6, and the expression of adhesion molecules involved in inflammatory interactions between leukocytes and endothelial cells [4]. Babcock et al. reported that ω-3 PUFAs, such as eicosapentaenoic acid and docosahexaenoic acid, significantly inhibit the production of TNF-α and IL-10 in monocytes [5]. The anti-inflammation effects of ω-3 eicosapentaenoic acid and the inflammation reactions of ω-6 arachidonic acid have been studied largely, but α-linolenic acid (a precursor of eicosapentaenoic acid) and linoleic acid (a precursor of arachidonic acid) had been not. In this study, to investigate the underlying mechanisms of C18 fatty acids on anti-inflammatory response, we measured the effect of C18 fatty acids such as stearic acid (C18, saturated fatty acid), oleic acid (C18:1: ω-9), linoleic acid (C18:2: ω-6) and α-linolenic acid (C18:3: ω-3) on intracellular Ca2+ mobilization and histamine release in RBL-2H3 mast cells.
METHODS
Materials
C18 fatty acids (stearic acid, oleic acid, linoleic acid and α-linolenic acid), verapamil, 8-(diethylamino)octyl-3,4,5-trimethoxybenzoate hydrochloride (TMB-8), bisindolylmaleimide (BIM), 3-(4,5-dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium bromide (MTT) and o-phthaldialdehyde (OPT) were obtained from Sigma Chemical (St. Louis, MO, USA). Fura-2/AM was purchased from Enzo Life Science (Farmingdale, NY, USA). [3H] Inositol was obtained from PerkinElmer (Waltham, MA, USA). AG 1-X8 resin was purchased from BIO-RAD (Hercules, CA, USA). The materials for cell culture were purchased from Life Technologies (Grand Island, NY, USA). Unless otherwise stated, all reagents were of the highest purity and were purchased from Sigma Chemical.
Cell culture
Rat basophilic leukemia (RBL)-2H3 cells were obtained from the American Type Culture Collection (ATCC, Manassas, VA, USA) and cultured as recommended by ATCC at 37℃ in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) and antibiotics (100 U/mL penicillin G, 100 µg/mL streptomycin) in a 5% CO2 incubator. Cells between 5 and 20 passages were used for experiments at a density of 1×106 cells/mL.
Measurement of cell viability
Cell viability testing was performed using a MTT-based colorimetric assay [6]. Cells were diluted and cultured in 96-well plates (1×104 cells/mL), with the select C18 fatty acid added (100~200 µM). An equal amount of ethanol was added in the control group, which resulted in a final concentration of 0.1% in the culture medium. MTT was added to the medium to a final concentration of 200 µg/mL and incubated for another 4 h at 37℃. After removing the culture medium, 200 µL of dimethylsulfoxide was added to the cells to dissolve the formazan. The absorbance of each well was measured by using a FlexStation device (Molecular Devices, Sunnyvale, CA, USA) at wavelength of 520 nm [7].
Measurement of intracellular Ca2+ mobilization
The intracellular Ca2+ level was measured using Fura-2/AM by monitoring a fluorospectrometer [8]. Briefly, culture medium was replaced and cells were washed three times with phosphate buffered saline (PBS). Cells were detached using 0.5% trypsin/EDTA buffer and suspended in 10 mL Krebs buffer [120.8 mM NaCl, 4.5 mM KCl, 1.2 mM MgCl2, 1.8 mM CaCl2, 5.6 mM glucose, 1.2 mM NaH2PO4 and 25 mM HEPES (pH 7.4)]. Fura-2/AM was added to a final concentration of 2 µM and incubated at 37℃ for 1 h. The cells were washed twice with Krebs buffer and centrifuged at 3,000×g for 10 min. For the experiments performed in the absence of external calcium, Krebs-EGTA buffer [120.8 mM NaCl, 4.5 mM KCl, 1.2 mM MgCl2, 1.0 mM EGTA, 5.6 mM glucose, 1.2 mM NaH2PO4 and 25 mM HEPES (pH 7.4)] was used. Fura-2 fluorescence was monitored by a Quanta Master device (Photon Technology International, Birmingham, NJ, USA.) at 37℃ with excitation at 340 and 380 nm and emission at 500 nm. The ratio of F340/F380 was recorded, and the maximum fluorescence ratio (Rmax) was measured using 0.1% Triton X-100. The minimum fluorescence ratio (Rmin) was measured following depletion of external Ca2+ by adding 5 mM EGTA/Tris, pH 8.5 [9,10].
Measurement of [3H]inositol phosphates formation
The cells were labeled with [myo-3H]inositol (1 µCi/106 cells) for 24 h at 37℃ and rinsed twice with inositol-free medium containing 0.5% FBS, 20 mM of LiCl and bovine serum albumin (1 mg/mL) and then resuspended at a density of 2×107 cells/mL. A portion (1 mL) of the cell suspension was transferred to a microcentrifuge tube and incubated at 37℃ for 15 min. Phosphatidylinositol 4,5-bisphosphate (PIP2) hydrolysis was initiated by adding 100 µM fatty acids for 30 min. Reactions were terminated by adding 200 µL of ice-cold 10% perchloric acid (HClO4). After 30 min in an ice bath, the tubes were centrifuged and the supernatants were diluted 5-fold with distilled water and applied to Dowex AG 1-X8 anion exchange columns. Each column was washed with 2 mL of distilled water followed by 10 mL of 60 mM ammonium formate containing 5 mM sodium tetraborate. Total inositol phosphates were eluted with a solution containing 1 M ammonium formate and 0.1 M formic acid. The radioactivity of the [3H]inositol phosphates was determined using a Tri-Carb Liquid Scintillation Counter (PerkinElmer, Waltham, MA, USA) [11].
Histamine release assay
The method that the determination of histamine is based on the reaction of histamine with OPT, which results in a highly fluorescent condensation product [12]. The harvested RBL-2H3 cells were washed with Krebs buffer and suspended in Krebs buffer at a density of 106 cells/mL. The cells were treated with BIM for 10 min and histamine release was induced by C18 fatty acids for 30 min in 37℃. After centrifugation, histamine contents in both supernatant and pellet were measured with 0.1 mL of 1% OPT in methanol. After 4 min, the reaction was terminated by adding 0.2 mL of 3 N HCl. The fluorescence intensity was measured using excitation and emission wavelengths of 355 and 455 nm, respectively, with a FlexStation apparatus. Data are expressed as % release (histamine contents in supernatant / histamine contents in supernatant and pellet ×100).
RESULTS
Effects of C18 fatty acids on the viability of RBL 2H3 cells
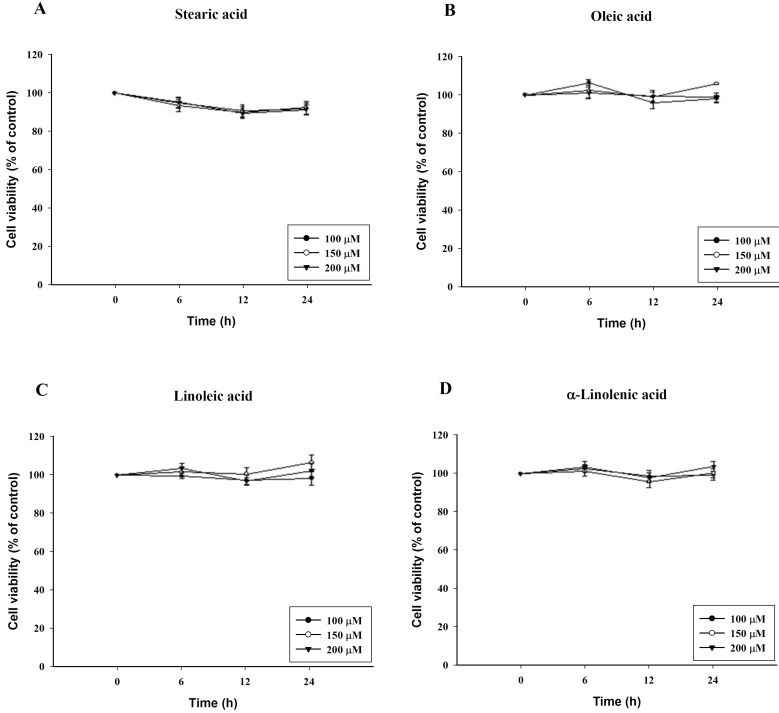
To confirm cytotoxicity of C18 fatty acids, cell viability was determined by the ability of the cells to metabolically reduce MTT to a formazan dye. It was performed after 6, 12 and 24 h exposure to the C18 fatty acid stearic acid, oleic acid, linoleic acid or α-linolenic acid. After 24 h exposure to oleic acid, linoleic acid and α-linolenic acid, which contain a double bond, no cytotoxicity was evident at a concentration of 200 µM. In contrast, stearic acid, which lacks a double bond, reduced the cell viability by 16% at 200 µM (Fig. 1). However, stearic acid showed no cytotoxicity at 6 h.
C18 fatty acid-induced intracellular Ca2+ mobilization
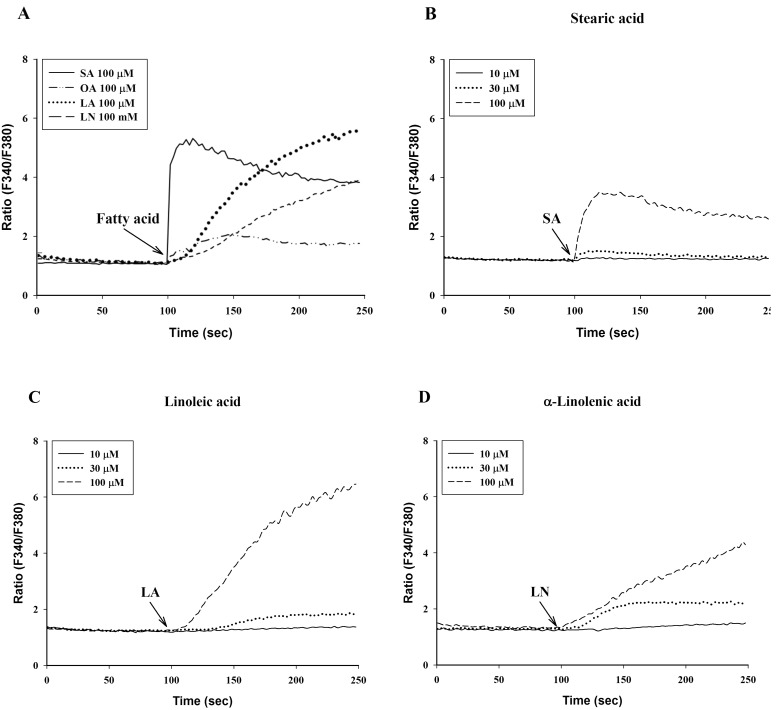
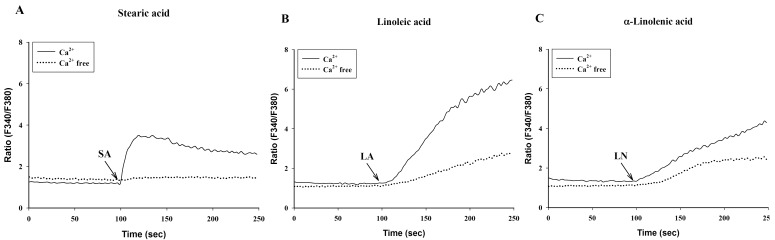
To investigate the effects of C18 fatty acids on intracellular Ca2+ concentration, we measured Ca2+ mobilization in Fura-2/AM-loaded RBL-2H3 cells. As shown in Fig. 2A, stearic acid rapidly increased the initial peak of intracellular Ca2+ mobilization, whereas linoleic acid and α-linolenic acid gradually increased intracellular Ca2+ mobilization. Oleic acid (100 µM) slightly increased intracellular Ca2+ concentration, which was smaller than the effects of other C18 fatty acids (Fig. 2A). In the next experiment, we excluded oleic acid. Stearic acid, linoleic acid and α-linolenic acid dose-dependently increased intracellular Ca2+ mobilization (Figs. 2B-2D). In the absence of extracellular Ca2+, stearic acid (100 µM) did not cause any increase of intracellular Ca2+ mobilization, but both linoleic acid and α-linolenic acid increased intracellular Ca2+ mobilization; the increase was smaller than that in the presence of extracellular Ca2+ (Fig. 3). These data suggest that C18 fatty acid-induced intracellular Ca2+ mobilization is mainly dependent on extracellular calcium influx.
Effects of calcium antagonists and BIM on C18 fatty acid-induced intracellular Ca2+ mobilization
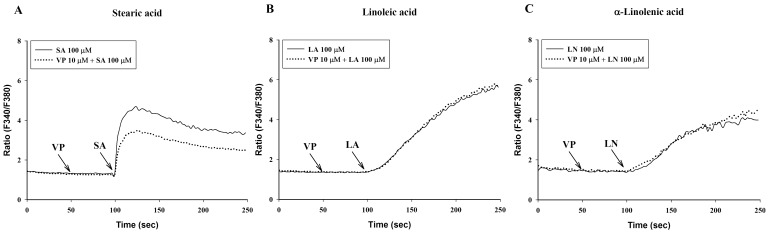
Verapamil (10 µM), a voltage-dependent calcium channel blocker, dose-dependently inhibited stearic acid-induced intracellular Ca2+ mobilization (Figs. 4A and 5), but did not affect linoleic acid- and α-linolenic acid-induced intracellular Ca2+ mobilization (Figs. 4B and 4C). At 10 µM TMB-8, a intracellular calcium release blocker, did not influence intracellular Ca2+ mobilization induced by stearic acid, linoleic acid and α-linolenic acid (data not shown). At 10 µM, BIM, a protein kinase C (PKC) inhibitor, significantly inhibited both linoleic acid- and α-linolenic acid-induced intracellular Ca2+ mobilization, but did not affect stearic acid-induced intracellular Ca2+ mobilization (Fig. 6).
Effects of C18 fatty acids on [3H]inositol phosphates formation in RBL-2H3 cells
The phospholipase C (PLC) pathway is closely involved in the intracellular Ca2+ mobilization. In [3H]inositol-labeled cells, melittin as a positive control significantly increased [3H]inositol phosphate formation by 245%. However, three C18 fatty acids did not increase [3H]inositol phosphate formation (Table 1), indicating that C18 fatty acids-induced intracellular Ca2+ mobilization is not associated with intracellular Ca2+ release by the PLC pathway.
Effects of C18 fatty acids on histamine release in RBL-2H3 cells
At 100 µM, linoleic acid and α-linolenic acid significantly increased histamine release from 15.2±4.8% (control) to 54.3±7.7% and 44.2±8.8%, respectively, which was smaller than the effect of 100 µM arachidonic acid (69.7±2.4%). However, stearic acid, which lacks a double bond, did not induce histamine release at a concentration of 100 µM (Fig. 7A). At 10 µM, the BIM, PKC inhibitor, significantly inhibited linoleic acid- and α-linolenic acid-induced histamine release (Fig. 7B), suggesting the involvement of PKC in linoleic acid- and α-linolenic acid-induced histamine release in RBL-2H3 cells.
DISCUSSION
In the measurement of histamine release, it is important to confirm the cytotoxicity of C18 fatty acids. To measure histamine release from RBL-2H3 cells, C18 fatty acids were exposed to the cells for 1 h. This exposure was not toxic, since no cytotoxicity was apparent after 6 h exposure of the tested fatty acids. These data suggest that exocytosis of histamine may be independent on cell membrane rupture. It has been reported that exposure of stearic acid at 150 µM for 24 h induces cell death and that ω-6 PUFAs have a greater protective effect on the deleterious effect caused by stearic acid in ECV-304 endothelial cells than ω-3 PUFAs [13].
Stearic acid rapidly increased the initial peak of intracellular Ca2+ mobilization, whereas linoleic acid and α-linolenic acid gradually increased intracellular Ca2+ mobilization. In the absence of extracellular Ca2+, stearic acid (100 µM) did not cause any increase of intracellular Ca2+ mobilization. Both linoleic acid and α-linolenic acid increased intracellular Ca2+ mobilization, but the increase was smaller than that in the presence of extracellular Ca2+. These results suggest that C18 fatty acid-induced intracellular Ca2+ mobilization is mainly dependent on extracellular calcium influx. A prior study reported that 40 µM of three ω-6 polyunsaturated fatty acids (arachidonic acid, γ-linolenic acid and linoleic acid) increased intracellular calcium concentration, while 40 µM ω-3 and ω-9 PUFAs did not [14]. The discrepancy between the present and previous study may be due to the concentration of C18 fatty acids because we used 100 µM of the C18 fatty acids.
At 10 µM, verapamil, a voltage-dependent calcium channel blocker [15], inhibited stearic acid-induced intracellular Ca2+ mobilization, but did not affect both linoleic acid and α-linolenic acid-induced intracellular Ca2+ mobilization. The PKC inhibitor BIM used at 10 µM significantly inhibited both linoleic acid and α-linolenic acid-induced intracellular Ca2+ mobilization, but it did not affect stearic acid-induced intracellular Ca2+ mobilization. These data suggest that the underlying mechanism of stearic acid, linoleic acid and α-linolenic acid on intracellular Ca2+ mobilization may be different from each other. Also, C18 fatty acids-induced intracellular Ca2+ mobilization appeared not to be associated with intracellular Ca2+ release by the PLC pathway, since they failed to induce [3H]inositol phosphates formation in [3H]inositol-labeled cells. This finding is consistent with a previous report that 40 µM arachidonic acid did not result in the formation of inositol-1,4,5-triphosphates [14]. Long chain fatty acids and ω-3 PUFAs (EPA and DHA) have been demonstrated to act as ligands of several G-protein-coupled receptors, such as GRP40 and GRP120, respectively [16,17]. Especially GRP40 activation which is strongly expressed in the pancreas results in an increase of intracellular Ca2+ mobilization via the phosphoinositide pathway. Further study will be needed to elucidate novel physiological roles of GRP40 and GRP120 in RBL-2H3 cells.
Clinical studies [18,19,20] indicated that the ingested ratio of ω-6 to ω-3 (especially linoleic vs. α-linolenic) fatty acids is important for maintaining cardiovascular health. While ω-3 PUFAs are extremely beneficial in preventing heart disease in humans, the levels of ω-6 PUFAs are insignificant [21,22]. Both ω-3 α-linolenic acid and ω-6 linoleic acid are essential fatty acids in humans. ω-3 and ω-6 C18 PUFAs compete for the same metabolic enzymes; thus, the ω-6: ω-3 ratio will significantly influence the ratio of the ensuing eicosanoids (hormones) including prostaglandins, leukotrienes and thromboxanes, and will alter the body's metabolic function [23]. Especially, linoleic acid (C18:2: ω-6)-induced intracellular Ca2+ mobilization and histamine release are more prominent than α-linolenic acid (C18:3: ω-3). These data support the view that increased intake of α-linolenic acid compared to linoleic acid is useful in preventing inflammation.
 XML Download
XML Download