

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Influenza causes worldwide outbreaks and occasional pandemics of acute respiratory disease every year [1]. Although usually self-limiting, influenza infection can pose a high morbidity and mortality to infants, the elderly, and individuals with underlying chronic diseases [2,3]. Influenza A viruses are enveloped negative-strand RNA viruses with eight single-stranded RNA segments encoding at least eleven viral proteins. The surface glyco-proteins hemagglutinin (HA) and neuraminidase (NA) embedded in the lipid bilayer of the viral envelope, are essential during viral replication and are the major antigenic determinants. The M1 protein is a matrix protein of the influenza virus. It forms a coat inside the viral envelope [4]. Although vaccines are available for influenza diseases, the compounding issue of antigen-dependent vaccination is antigenic shifts. To address this, influenza vaccines are reviewed annually to ensure protection is maintained [5]. Moreover, control of influenza is particularly important at the beginning of pandemic events [6], while antibodies and specific T cells are first detected around four days after infection [7]. Exposure to viral infections often occurs before antigen-dependent vaccines can provide optimal protection. The innate immune system which is presently recognized as an essential response to viral infection has no time lag and antigen specific recognition. It gives the host time to activate virus-specific adaptive immune responses that are needed for the clearance of the virus [8].
Toll-like receptors (TLRs) which are evolutionarily conserved innate receptors expressed in various immune and non-immune cells of the mammalian host, play a crucial role in defending against pathogenic microbial infection. An alternative strategy to defense against the influenza viruses may be to stimulate the innate immune response through the activation of TLRs signaling pathways [9,10]. Synthetic unmethylated CpG-containing oligodeoxynucleotide motifs (CpG ODNs) have been identified as specific ligands for TLR9. Their ability to stimulate Th1-like responses makes them suitable adjuvants for vaccines against viruses [11]. Pre-treatment of mice with CpG-ODN (TLR-9 agonist) have been found to provide complete protection against lethal seasonal influenza A/PR/8/34 (H1N1) [12]. The prior incorporation of CpG-ODN into the avian influenza virus (AIV) H5N1 oil emulsion vaccine activates both the humoral and Th1 type immune responses in chickens [13]. TLR-3 is expressed on respiratory epithelium and appears to play a central role in mediating both the antiviral and inflammatory responses of the innate immunity in combating viral infections [14,15]. Poly I:C is TLR-3 agonist and is potent inducer of interferon. Intranasal pretreatment of mice with Poly ICLC and LE Poly ICLC provides high level of protection against lethal challenge with a highly lethal avian H5N1 influenza [12].
Viral infection rapidly induces a cascade of interferons (IFNs) and other pro-inflammatory cytokines in vertebrate cells [16]. Type I interferons (IFN α/β) are the key cytokines produced predominantly by innate immune cells to combat viral infections [17]. Experiments using IFN-β knock-out mice have demonstrated the susceptibility to influenza virus [18]. Cytokines such as interleukin (IL)-6 and IL-12 play critical roles in amplifying airway inflammation caused by viral infection. Studies on influenza clinic volunteer have shown a direct correlation between disease severity, viral replication in the upper respiratory tract, and the levels of cytokines in nasal wash fluids and plasma [19,20].
Epithelial cells, the first defense barrier against virus in human tissues, potentially affect host defense against influenza viruses. Studies on the respiratory epithelial cells response to influenza virus have not been widely investigated and remain controversial. In this study, we investigated the innate immune response against influenza virus in an in vitro model of human pharyngeal epithelial cells (Hep-2). We also analyzed for the antiviral potency of two TLR agonists in inducing protective antiviral immunity against influenza viruses in Hep-2 cells. These findings will provide important information for new drug design and development for the treatment of influenza virus.
Go to : 
METHODS
Cells and virus
Human pharyngeal epithelial cells (Hep-2) were cultured in Minimum Essential Medium with Earle's Balanced Salts and L-glutamine (MEM-EBSS) supplemented with 5% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 µg/ml streptomycin at 37℃ in a humidified atmosphere of 5% CO2. The culture medium was replaced every two days.
An influenza virus stock was used of a virus isolate which was obtained during the influenza epidemic 2008 in Liaoning, A/Liaoning/1136/2009 (sH1N1). The preparation of this virus stock has been described previously [21]. Briefly, the virus was propagated in Madin-Darby canine kidney (MDCK) cells to make working stocks of the viruses in serum-free Dulbecco's Modified Eagles Medium (DMEM) supplemented with tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-treated trypsin (2 µg/ml) at 33.5℃ under 5% CO2. When 100% cytopathogenic effect (CPE) was achieved, cell debris was removed by centrifugation and the infected culture fluid was stored in aliquots at -80℃. Viral titers were expressed as multiplicity of infection (moi) based on MDCK plaque forming units. All cell culture reagents were obtained from Gibco BRL.
Infection of Hep-2 cells with influenza virus
Hep-2 cells were plated overnight in culture medium in 24-well plates at a density of 100,000 cells per well. Cells were washed twice with PBS, exposed to sH1N1 at a moi of 1.0 or 0.01. On each plate a control well (no virus) was included. Virus was allowed to be adsorbed by the cells for 1 hr with gentle rocking of the plates every 20 min. After 1 hr, virus was removed and 1 ml of serum-free MEM-EBSS medium with 100 U/ml penicillin, 100 µg/ml streptomycin and trypsin (1.5 µg/ml) (Gibco BRL) was added. Infected cells were incubated at 37℃ under 5% CO2 in humidified air. At t=1, 8, 24, 48, 72 and 96 hr post infection, culture supernatants were discarded and cells were collected for RNA isolation at each time point.
Stimulation and infection protocol
The sequences of unmethylated CpG-ODN used in the present study was: GTC GTT GTC GTT GTC GTT [22], synthesized by TaKaRa. The synthetic dsRNA analog, poly I:C, was obtained from InvivoGen (San Diego, CA, USA). Hep-2 cells were seeded over night into 24-well plates containing 100,000 cells per well in 1ml of complete MEM-EBSS medium at 37℃ under 5% CO2. Prior to stimulation, cells were washed once more with PBS and then stimulated with CpG-ODN (5 µM), poly I:C (25 µg/mL), or a combination of the two at 37℃ under 5% CO2. After 24 hr the conditioned medium was removed, cells were washed with PBS and infected with sH1N1 at a moi of 1.0 or 0.01 in MEM-EBSS medium without FBS for 1 hr with gentle rocking of the plates every 20 min at 37℃ under 5% CO2. Next, the sH1N1 was removed and replaced by serum-free MEM-EBSS medium with 100 U/ml penicillin, 100 µg/mL streptomycin and trypsin (1.5 µg/ml). At t=48 (moi 1.0) or 72 hr (moi 0.01) post infection, culture supernatants were discarded and cells were collected for RNA isolation. Cells which were simultaneously stimulated with PBS and exposed to virus were used as a control.
RNA isolation, cDNA synthesis, and RT-PCR
Total RNA was isolated from frozen cell pellets using RNeasy Mini Kit (Qiagen) according to the manufacturer's instructions. Isolated total RNA was eluted in RNase-free water and stored at -80℃ until use. Reverse transcription (RT) was performed using the PrimeScript™ RT reagent Kit with gDNA Eraser (TaKaRa, Dalian, China), according to the manufacturer's instructions. Briefly, the mixture of total RNA (2 µl), 5× gDNA Eraser Buffer (2 µl), gDNA Eraser (1 µl) and RNase-free dH2O (5 µl) was incubated at 42℃ for 2 min to eliminate genomic DNA. After a brief centrifugation, the mixture was mixed with 5× PrimeScript Buffer 2 (4 µl), PrimeScript RT Enzyme Mix (1 µl), and Rnase-free dH2O to total volume of 20 µl. Single-strand cDNA was synthesized at 37℃ for 15 min and 85℃ for 5 s in an ABI Veriti thermal cycler (Applied Biosystems, USA) and stored at -80℃.
Polymerase chain reactions (PCR) were performed according to standard protocols with the primers indicated in Table 1. PCR conditions were as follows: 95℃ for 30 s, followed by 40 cycles of 95℃ for 5 s, 60℃ for 34 s. PCR products were visualised on a QIAxcel instrument (Qiagen) using QIAxcel ScreenGel Software.
Quantitative real-time reverse transcription PCR (qRT-PCR)
Real-time quantitative PCR was performed as described in SYBR Premix Ex Taq™ II (TaKaRa, Dalian, China). Briefly, 2 µl of cDNA was combined with SYBR Premix Ex Taq™ II, 0.8 µl of primers (10 µM), 0.4 µl of ROX Reference Dye II (50×) and RNase-free water to total volume of 20 µl. The cycling protocol was 95℃ for 30 s, followed by 40 cycles of 95℃ for 5 s, 60℃ for 34 s, and a dissociation stage of 95℃ for 15 s, 60℃ for 1 min, 95℃ for 15 s and 60℃ for 15 s in an ABI 7500 Real-Time PCR System (Applied Biosystems, USA). The generation of a specific PCR product was tested using melting curve analysis. Primer sequences used for qRT-PCR can be found in Table 1.
The amounts of TLRs and cytokines mRNA relative to unstimulated cells were calculated with 2-ΔΔCt method and the mRNA levels were normalized against GAPDH mRNA. Gene slopes were determined with a series of 10-fold RNA dilutions (data not show).
To determine the actual number of sH1N1 copies, a reference standard curve was used. Dilutions were made from a plasmid, which contains the sH1N1 PCR-target sequence. Ten-fold dilutions equivalent to 1×101 to 1×106 copies per reaction were prepared.
Statistical analysis
Data are means and standard deviation from three independent experiments. Data were analyzed using one-way analysis of variance or paired-sample t tests. p<0.05 was considered to be significant.
Go to :
RESULTS
Influenza virus replication in Hep-2 cells
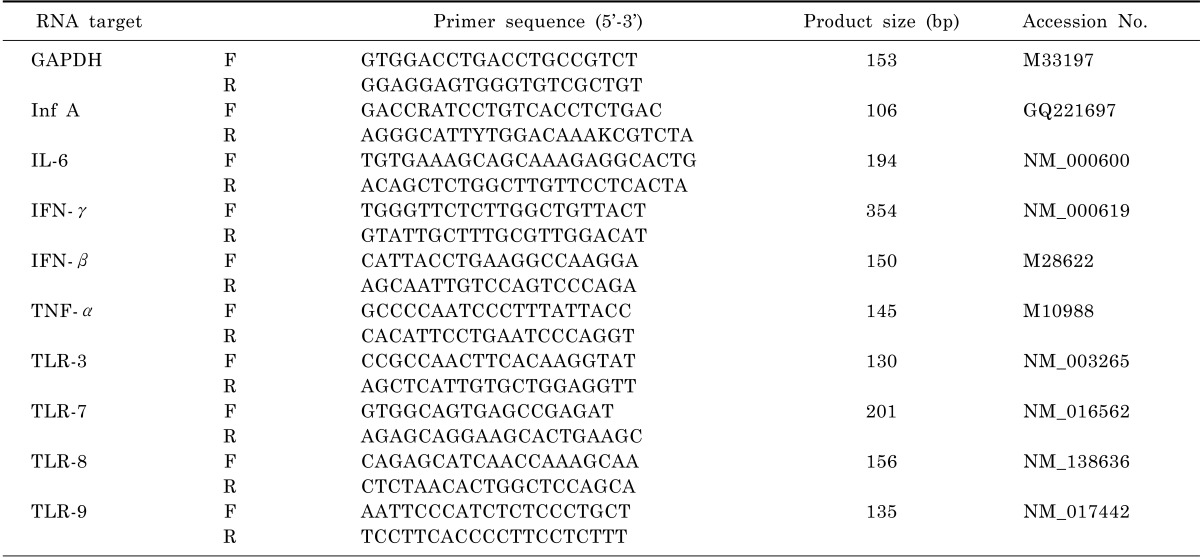
The most sensitive method for monitoring influenza virus replication in vitro is qRT-PCR assay. To determine virus dose effects, we infected Hep-2 cells with sH1N1 virus at a moi of 1.0 and 0.01. As shown in Fig. 1, at a moi of 1.0, a rise in viral load was observed at 8 hr post infection, reaching a maximum of 8.57×107 copies/mL 24 hr post infection. Also at a moi of 0.01, viral replication was observed at 8 hr post infection, while the peak of 8.06×107 copies/ml was reached 48 hr post infection, 24 hr later than for a moi of 1.0. When Hep-2 cells were infected without addition of trypsin, viral replication could not be detected at any time point (Data not shown).
Human pharyngeal epithelial cells in culture constitutively expressed TLRs and proinflammatory cytokines
To determine TLRs and proinflammatory cytokines mRNA distribution in Hep-2 cells, total RNA was isolated from vehicle-only (without virus) treated Hep-2 cell at each time points, and subjected to RT-PCR analysis. The results conformed well to our expectations that the level of TLRs and proinflammatory cytokines expression were not varied among the different time points (a representative data is shown as Fig. 2). For TLRs, TLR3 and TLR9 were detected at moderate levels, whereas TLR7 and TLR8 were not detected (Fig. 2). For proinflammatory cytokines, IL-6 transcripts were most abundant, followed by IFN-β, TNF-α was expressed at very low but detectable levels, IFN-γ was not detected.
 | Fig. 2Expressions and distribution of TLRs and proinflammatory cytokines in Hep-2 cell lines. Total RNA was isolated from vehicle-only (without virus) treated Hep-2 cells and subjected to RT-PCR using primers as indicated in Table 1. The amplification products were estimated by capillary electrophoresis on a Qiaxcel apparatus (Qiagen).
|
Influenza virus upregulate the TLRs in Hep-2 cells
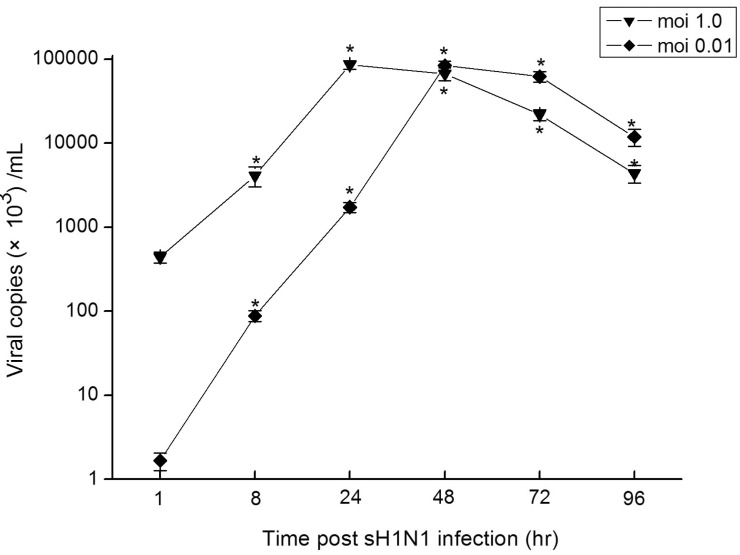
To identify whether TLR3 and TLR9 expressed in these epithelial cells actually functional upon stimulation with the virus, we analyzed the time course of TLR3 and TLR9 expressions in Hep-2 cells in response to sH1N1 virus. As Fig. 3 shows, the influenza virus caused a significant induction of both TLR3 and TLR9 in Hep-2 cells. At a moi 1.0, the peak induction of TLR3 and TLR9 by sH1N1 virus occurred at 48 hr post-infection, 9 and 12-folds respectively over the control. When Hep-2 cells were infected at a moi of 0.01, the peak induction of TLR3 and TLR9 were reached 72 hr post-infection, 24 hr later than for a moi of 1.0 (Fig. 3). These findings indicate that TLR3 and TLR9 in Hep-2 cells might be functional receptors in response to sH1N1 virus infection.
 | Fig. 3Time course of TLRs expressions in Hep-2 cells in response to sH1N1 virus. Hep-2 cells were exposed to sH1N1 virus at a moi 1.0 or 0.01 for 1, 8, 24, 48, 72 or 96 hr and the expression of TLR3 (a) and TLR9 (b) mRNA were then determined by qRT-PCR. Transcript levels normalized to GAPDH and expressed relative to unstimulated cells. Data are the averages±S.D. of triplicate determinations.
|
Secretion of inflammatory cytokines from influenza virus-infected Hep-2 cells
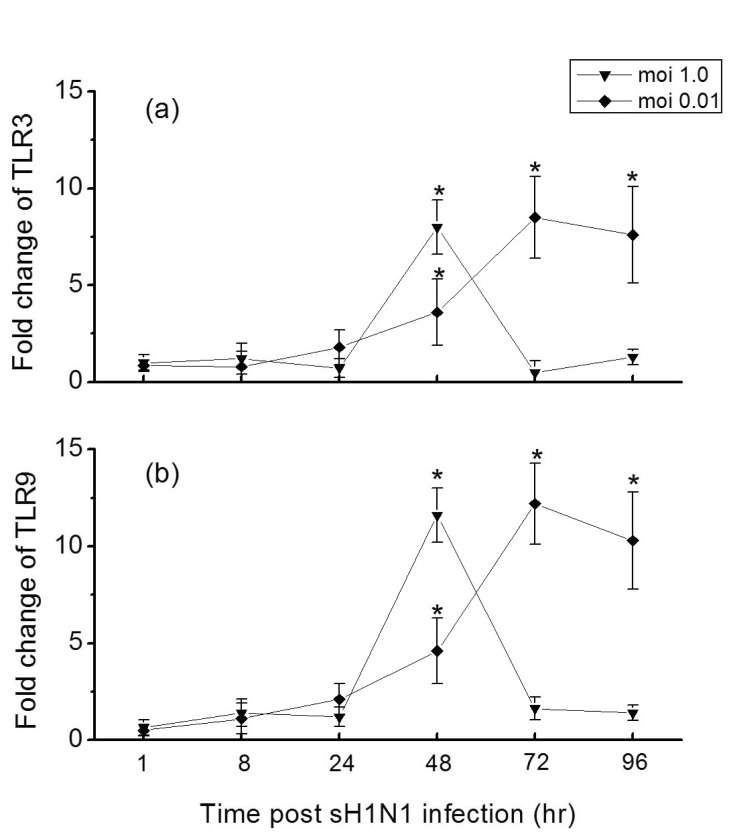
Since activation of TLRs may induce or amplify cytokines production, we sought to further examine the capacity of influenza virus to induce secretion of cytokines in Hep-2 cells. As Fig. 4 shows, influenza virus elicited a rapid and significant increase of IL-6, IFN-β, and TNF-α mRNA expression in Hep-2 cells. At a moi 1.0, the mRNA levels of these three cytokines reached the maxima at 48 hr post infection, while at a moi of 0.01, the peak induction of them were observed at later time points (t=72 hr). Depending on the moi, the level of IL-6 mRNA increased by 212-fold in Hep-2 cells at a moi of 1.0, markedly higher than for a moi of 0.01 (83-fold) (Fig. 4a). Not as IL-6, the levels of IFN-β and TNF-α mRNA induced from cells infected at a moi of 1.0 or 0.01 were similar (Fig. 4b and 4c).
 | Fig. 4Time course of cytokines expressions in Hep-2 cells in response to sH1N1 virus. Hep-2 cells were exposed to sH1N1 virus at a moi 1.0 or 0.01 for 1, 8, 24, 48, 72 or 96 hr and the expression of IL-6 (a), IFN-β (b) and TNF-α (c) mRNA were then determined by qRT-PCR. Transcript levels normalized to GAPDH and expressed relative to unstimulated cells. Data are the averages±S.D. of triplicate determinations.
|
Antiviral potency of TLRs ligands
To identify any inhibitory effect of TLRs agonists on influenza virus replication, the matrix protein 1 (M1) mRNA of influenza virus A was compared between TLRs agonist treated and untreated infected cells. Hep-2 cells were pre-incubated with CpG-ODN (5 µM), poly I:C (25 µg/mL) alone or in combinations of two for 24 hr and then exposed to sH1N1 virus. Total RNA extraction was performed at 48 hr after influenza virus infection, and the levels of intracellular influenza mRNA were measured. As illustrated in Fig. 5, at infection of moi 1.0, no reduction in viral mRNA expression was observed when cells were pre-treated with poly I:C, CpG-ODN alone or combination of the two. In contrast, at moi 0.01, Hep-2 cells produced a significant reduction in viral mRNA expression when stimulated with either CpG-ODN or poly I:C alone. The combination of the two TLR agonists did not demonstrate a synergistic or additive affect on viral mRNA reduction.
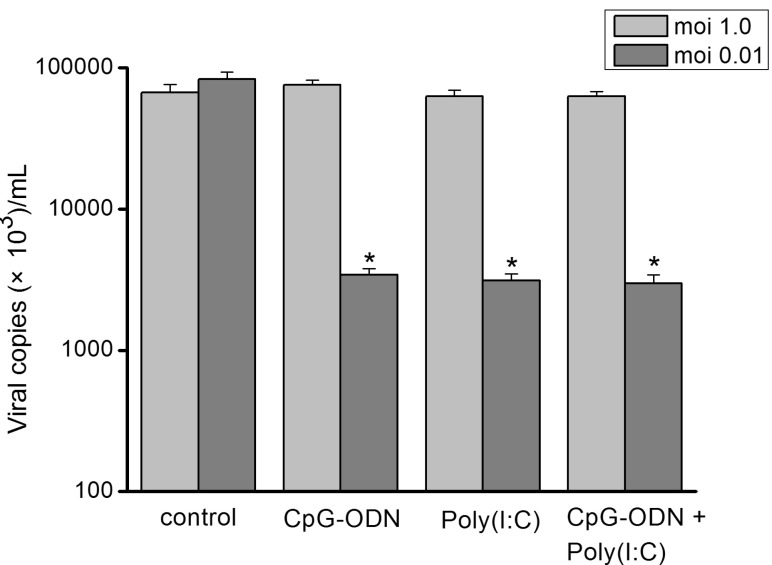 | Fig. 5Differential reduction of viral loads by TLRs ligands. Hep-2 cells were pre-incubated with CpG-ODN (5 µM), poly I:C (25 µg/ml) or in combinations of two for 24 hr and exposed to sH1N1 virus at a moi 1.0 or moi 0.01 for additional 48 hr. Cells which were simultaneously pre-incubated with PBS and exposed to sH1N1 virus at a moi 1.0 or moi 0.01 for additional 48 hr was used as a control. Viral loads were determined using qRT-PCR. Data are the averages±S.D. of triplicate determinations (*p<0.05 compared with control).
|
TLR ligands influence on cytokine production
The virus-induced 'cytokine storm' appears to contribute to the influenza viruses pandemics [23,24]. In addition to investigating inhibitory effects against the virus, the influence of TLRs ligands on the sH1N1-induced cytokines were further studied. Hep-2 cells were pre-incubated with CpG-ODN (5 µM), poly I:C (25 µg/ml) alone or in combinations of two for 24 hr prior to addition of sH1N1 virus at a moi 1.0 or moi 0.01 for additional 48 hr or 72 hr, respectively. The levels of intracellular cytokines expression were compared to cells which were simultaneously stimulated with PBS plus sH1N1. At infection of moi 1.0, CpG-ODN and poly I:C induced a small, though significant decrease in cytokines mRNA expression. In contrast, at moi 0.01, pretreatment of Hep-2 cells with CpG-ODN or poly I:C almost completely suppressed the cytokines elevation. Combination of the two TLR ligands had no additive effect compared with the polyI:C or CpG-ODN alone (Fig. 6).
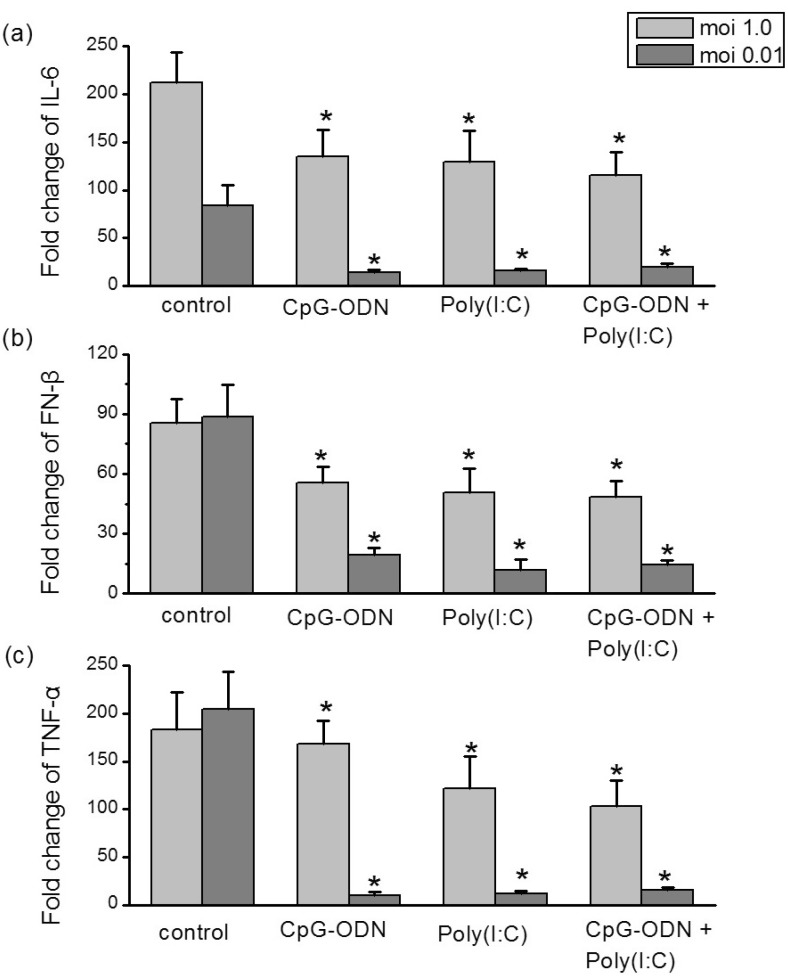 | Fig. 6Differential reduction of cytokines mRNA levels by TLRs ligands. Hep-2 cells were pre-incubated with CpG-ODN (5 µM), poly I:C (25 µg/ml) or in combinations of two for 24 hr and exposed to sH1N1 virus at a moi 1.0 or moi 0.01 for additional 48 hr or 72 hr, respectively. Cells which were simultaneously pre-incubated with PBS and exposed to sH1N1 virus at a moi 1.0 or moi 0.01 for additional 48 hr or 72 hr, respectively, was used as a control. IL-6 (a), IFN-β (b) and TNF-α (c) mRNA expression levels were determined using qRT-PCR. Transcript levels normalized to GAPDH and expressed relative to unstimulated cells. Data are the averages±S.D. of triplicate determinations (*p<0.05 compared with control).
|
Go to :
DISCUSSION
For sH1N1, Li et al. recently demonstrated that viral load of ≥106 copies/ml was detected only in human cell lines derived from tissues of lower respiratory tract, gastrointestinal tract, liver, kidney and muscle. CPE was present only in 4 cell lines for sH1N1 (A549, Caco-2, HeLa, HEK) [25]. In this study we have demonstrated that sH1N1 replicated to 8×107 copies/ml in Hep-2 cells (Fig. 1), suggesting its capability to replicate in upper respiratory tract cell lines. Moreover, when Hep-2 cells were infected without addition of trypsin, viral replication could not be detected at any time point (Data not shown). After the addition of exogenous trypsin to Hep-2 cell lines, the viral load of sH1N1 increased by at least 2 log10 copies/ml. Presence of multiple basic amino acids at the cleavage site of haemagglutinin precursor molecule, which can be cleaved by ubiquitous proteases, allows efficient replication in body sites with trypsin and non-trypsin endogenous proteases [26]. In the present study, Though, >2 µg/ml trypsin induced degeneration in Hep-2 cell lines, 1.0~1.5 µg/ml trypsin was required for the cleavage of haemagglutinin precursor, and therefore increased viral replication (Data not shown).
Toll-like receptors serve as pattern recognition receptors (PRRs) by initiating signals that elicit anti-viral immune responses. When activated, they trigger immune and inflammatory responses to respond to the invading microorganism. There are four known toll-like receptors found in mammalians cells which recognize nucleic acids. They are TLR-3, TLR-7, TLR-8 and TLR-9 [27]. In this study, we found clear expressions of TLR3 and TLR9 in Hep-2 cells using RT-PCR (Fig. 2). After sH1N1 infection, expression of the TLR3 and TLR9 were significantly up-regulated (Fig. 3), which indicated that TLR3 and TLR9 in Hep-2 cells might be functional receptors in inducing anti-viral responses in general.
It has been suggested that synthetic ligands, targeting TLR receptors, may have a therapeutic potential as antiviral compounds [28]. Therefore, in this study the antiviral potency of Poly I:C and CpG-ODN, which were agonists specific for TLR3 and TLR9 respectively, were examined in an in vitro model of sH1N1 infection. Our data showed, pretreatment with CpG-ODN or poly(I:C) alone significantly reduced viral replication in Hep-2 cells at lower viral challenge doses (moi=0.01). While, at higher viral challenge doses (moi=1.0), the agonists treatment did not work effectively. Not as expected, the synergy in limiting sH1N1 replication was not observed when the combinations of CpG-ODN (5 µM), poly I:C (25 µg/ml) were used (Fig. 5). These results demonstrate that TLR3 and TLR9 pathways may contribute to the observed antiviral effects, while the effects of them were rather limited.
Type I IFNs are the most important antiviral cytokines produced by cells in response to viral infection [9]. TNF-α has also been shown to have anti-influenza virus effect in lung epithelial cells [29]. In this study, we observed a strong induction of IFN-β and TNF-α in influenza-infected Hep-2 cells (Fig. 4b and 4c), suggesting IFN-β and TNF-α have important functions at early times of virus infection. Influenza infection triggers a cascade of pro-inflammatory cytokines that is responsible for virus control and clearance, but also for many of the symptoms associated with the disease [30]. Herein, we found that the production of IL-6, which is considered indicative of severe disease in humans [31,32], was delivered dependent on the infecting viral dose (Fig. 4a). We hypothesize that the elevated production of IL-6 may simply represent a response to increased pathology. Furthermore, cytokines expressed in Hep-2 cells is consistent with the previously published findings from human volunteer studies and mouse experiments [33,34], demonstrating the value of the Hep-2 model for the study of the immune response to influenza.
Cytokine storms are associated with a wide variety of infectious and noninfectious diseases [35]. In the case of infections with H5N1, a highly pathogenic influenza strains, the cytokine response becomes dysregulated, resulting in a massive inflammatory response contributes to systemic tissue damage [23]. Therefore, the inhibition of virus-induced cytokine release is important for the treatment of influenza [36]. Although it is well recognized that certain TLR ligands are able to stimulate the release of cytokines, our results showed that TLR3 and TLR9 ligands (CpG-ODN and poly I:C) could significantly reduce sH1N1 virus-induced cytokine secretion. The reason for this discrepancy is not entirely clear, but could indicate that TLR ligands also possess an anti-sH1N1-induced-inflammation activity.
In summary, we have demonstrated that sH1N1 influenza viruses marginally increased the expression of TLRs and cytokines in Hep-2 cells. The ability of sH1N1 influenza viruses to modulate immune responses in Hep-2 cells may be a feature shared by the other virulent influenza viruses. Moreover, we demonstrated a strong antiviral effect of certain TLR ligands, which may represent an affective antiviral strategy to combat influenza viruses. The activity study of TLR ligands on influenza and underlying mechanisms will provide a theoretical and experimental basis to guide novel anti-influenza virus drug discovery.
Go to :
 XML Download
XML Download