

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Ion channels play critical roles during essential physiological functions, such as, muscle contraction and hormone secretion [1], and ion channel defects lead to a variety of diseases including cancer [2-5]. Several studies have recently revealed that transient receptor potential (TRP) channels are associated with cell proliferation, apoptosis, and cancer development [6-8]. These channels are non-selective cation channels (NSCCs), and initially, they were cloned from Drosophila melanogaster. All TRP channels possess putative six-transmembrane spanning domains. Based on their amino acid sequences, the TRP family can be divided into seven subfamilies, namely, TRPC (canonical), TRPM (melastatin), TRPA (ankyrin), TRPV (vanilloid), TRPP (polycistin), TRPN (NOMP-C homologues), and TRPML (mucolipin), which are activated by different physical and chemical stimuli. Furthermore, the various gating mechanisms of TRP channels play crucial roles in their pathologic and physiologic functions [9-14]. In this review, we mainly focus on current understanding of the molecular, biophysical, and pharmacological properties of TRPM7, on its physiological and pathophysiological roles, and on its therapeutic potential.
STRUCTURE
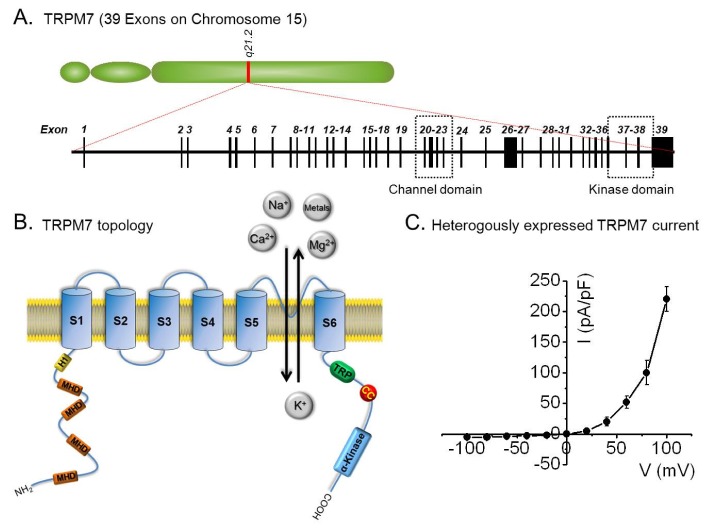
The TRPM7 gene is located on chromosome 15, and consists of 39 exons that encode an 1863-amino acid protein. To form a channel pore, TRPM7 is believed to form a tetrameric complex composed of four TRPM7 subunits. Each subunit is composed of six transmembrane segments (S1-6), and the loops of each subunit that form a channel pore are located between S5-S6. The E1047 and Y1049 residues in mouse TRPM7, within the putative pore loop, are critical for divalent cation selectivity [15-17], and the N-terminus of TRMP7 contains a hydrophobic region (H1) and four regions of the TRPM subfamily homology domain (MHD), but the biological significances of these regions remain largely undefined. Its C-terminus contains a TRP box of ~25 highly conserved residues, which may interact with phosphatidylinositol 4,5-bisphosphate (PIP2), a positive regulator of some TRP channels [9]. In addition, a coiled-coil (CC) domain close to the C-terminus may mediate subunit-subunit interactions and the tetrameric assembly of TRPM7 [18] (Fig. 1).
TRPM7 also contains an atypical serine/threonine protein kinase domain at its distal C-terminus, which is homologous to a family of α-kinases, and this feature is unique among ion channels [9,16,19]. Crystallographic studies of the TRPM7 kinase domain revealed that TRPM7 kinase consists of two lobes adjoining the nucleotide binding site, and structural similarity between its TRPM7 kinase domain and cAMP-dependent protein kinase A [16,20]. Although this kinase domain does not seem to affect channel activity directly [21,22], it may be important for the regulation of channel function by Mg2+ nucleotides [19,23].
BIOPHYSICAL PROPERTIES
TRPM7 currents exhibit a reversal potential of ~0 mV, and mediate small inward currents at negative physiological membrane potentials and pronounced outward rectification at positive membrane potentials. In the physiological range of -40 mV to -80 mV, TRPM7 conducts small inward currents by inwardly transporting divalent cations, such as, Ca2+ and Mg2+, from the extracellular space into the cytosol down their concentration gradients. On the other hand, at positive membrane potentials, TRPM7 outwardly transports intracellular cations, such as, K+, although at potentials exceeding +50 mV, outward monovalent cation fluxes become more significant [19]. This outwardly rectifying property is entirely due to a voltage-dependent block of monovalent cation influx by extracellular divalent cations. On the other hand, in the absence of external divalent cations, TRPM7 channels are permeable to monovalent cations and outward rectification is largely abolished [21].
TRPM7 is more permeable to trace metal ions than other Ca2+ permeable channels. Based on the results of their equimolar ion-substitution experiments, Monteilh-Zoller et al.[21] published a unique permeation profile order for TRPM7, as follows: Zn2+≈Ni2+≫Ba2+>Co2+>Mg2++Mg2++Mg2++Mg2++Mg2+. Fura-2 quenching experiments with Mn2+, Co2+, or Ni2+ showed that entry of these divalent ions is allowed by TRPM7 in the presence of physiological levels of Ca2+ and Mg2+ [21]. TRPM7 is constitutively activated and regulated by intracellular Mg2+ or Mg-ATP. Thus, TRPM7 currents are induced within several minutes by depleting internal Mg2+ using Mg2+-free pipette solutions or chelators in whole-cell configuration patch-clamp experiments [16]. TRPM7 have the dual ability to act simultaneously as a channel and as a kinase and the kinase domain is adjacent to the conducting pore of TRPM7, changes in free Mg2+ concentrations might activate the associated kinase domain, and thus, modulate TRPM7 channel gating [15,22,24,25]. TRPM7 is homologous to TRPM6 (a channel kinase); the two proteins exhibit 49% primary amino acid sequence identity. TRPM6 and TRPM7 have been characterized as Mg2+ and Ca2+ permeable ion channels essential for cellular Mg2+ homeostasis. Furthermore, both proteins share the unique feature of an ion channel fused to a kinase domain and shared homology with atypical α-kinases [15,26,27].
TRPM7 has important physiologic/pathologic functions, and therefore, is a viable therapeutic target. Thus, it is important that we understand the mechanisms engaged by specific TRPM7 blockers. Recent studies have demonstrated that the channel activities of TRPM7 can be modulated by various reagents. In particular, 2-APB (2-aminoe-thoxydiphenyl borate; a nonspecific modulator of several TRP channels) inhibits endogenously and heterologously expressed TRPM7 currents but potentiates TRPM6 currents at micromolar concentrations [27,28]. Thus, 2-APB can be used to differentiate TRPM6 and TRPM7 currents. TRPM7 is also blocked by carvacrol [29], 5-lipoxygenase inhibitors [30], nafamostat mesylate (a synthetic serine protease inhibitor) [31], Ca2+-activated small conductance K+ channel blocker NS8593 [32], waixenicin A (extracted from soft coral) [33], sphingosine and FTY720 [34]. Future studies will undoubtedly provide more information as to whether these exogenous modulators of TRPM7 are useful tools for determining how TRPM7 channel activity manipulation can be used therapeutically [34].
TRPM7 channel activity is regulated by extracellular pH. A decrease in extracellular pH strongly enhanced TRPM7 current activity in HEK-293 cells [35]. Moreover, protons might compete with Ca2+ and Mg2+ for binding sites, and thus, increase inward current by releasing the divalent cation block [36]. However, the TRPM7-like current in the FaDu cell line was insensitive to acidic conditions [37], whereas it was increased in HeLa cells (a human cervical epithelial) [38]. Although the effects of protons on endogenous TRPM7 remain controversial, it is known TRPM7 can be regulated by acidic pathophysiological conditions, including those associated with ischemic stroke [19].
TRPM7 REGULATION
Heterologously expressed and native TRPM7 channels appear to be constitutively active, and to be regulated positively or negatively by different modulators [39]. The most important regulators of TRPM7 channel activity are free Mg2+ and Mg-complexed nucleotides. Recent studies have demonstrated TRPM7 can be inhibited by Mg•ATP at physiological concentrations and that elevated Mg•ATP concentrations enhance the efficiency of TRPM7 channel by blocking free Mg2+ [22,39]. In addition, TRPM7 channel can be regulated by phospholipase C (PLC), which acts by cleaving phosphatidylinositol 4,5-bisphosphate (PIP2). The kinase domain of TRPM7 directly associates with the C2 domain of PLC, and activation of PLC results in PIP2 hydrolysis and a rapid decrease in recombinant TRPM7 channel activity under Mg2+-free conditions [24]. TRPM7 may also be associated with phosphorylation. A variant of TRPM7 with a missense mutation (T1482I) is found in a subset of patients with Guamanian amyotrophic lateral sclerosis (ALS-G) and Parkinsonism-dementia (PD-G) [22,40]. Based on computer analysis of secondary structures, Thr-1482 in fish, amphibians, birds, and primates, and Ser-1482 in rats and mice are potential substrates for autophosphorylation by the C-terminus serine/threonine α-kinase domain of TRPM7. However, the Ile-1482 mutation found in these patients cannot be phosphorylated. On the other hand, when recombinant TRPM7 with the T1482I mutation was heterologously expressed in HEK-293 cells, TMEP7 channels remained functional but showed increased sensitivity to Mg2+ inhibition and reduced phosphorylation as compared with the wild-type [19,22,40].
PHYSIOLOGICAL FUNCTION
TRPM7 channel is ubiquitously expressed in almost all tissues [41,42], although TRPM7 mRNA is expression is greatest in heart, the pituitary gland, bone, and adipose tissue [41]. Several authors have suggested that TRPM7 is closely associated with cellular growth and development under physiological conditions [22,43-45]. For example, loss of TRPM7 function was found to induce growth arrest and cell death in DT-40 B cells, because the coordination between cellular energy metabolism and Ca2+ and Mg2+ homeostasis was disrupted [46]. In addition, knockdown by TRPM7 specific small interfering RNA (siRNA) in human osteoblastic cells prevented Ca2+ and Mg2+ influx and inhibited cell proliferation [47]. In another study, the global deletion of TRPM7 in mice resulted in embryonic lethality and the tissue-specific deletion of TRPM7 in thymocytes disrupted thymopoiesis [48]. However, in thymocytes, total cellular Mg2+ levels were maintained after TRPM7 deletion, whereas the zebrafish TRPM7 mutant touchtone/nutria exhibited severe growth retardation and gross alterations in skeletal development [45]. In a recent functional analysis of TRPM7 in the xenopus embryo, it was concluded that TRPM7 is essential for gastrulation during vertebrate embryogenesis [49]. Interestingly, gastrulation defects caused by TRPM7 depletion can be rescued by Mg2+ supplementation or by an inactivating mutation within its kinase domain, but not in its channel domain [49]. In addition, it has been reported that heterozygous TRPM7 kinase-deficient mice remain viable, whereas homozygous TRPM7 kinase-deficient mice exhibit early embryonic lethality [50].
Several studies have suggested that TRPM7 also plays a role in cell adhesion. The overexpression of TRPM7 in HEK-293 cells was found to lead to cell rounding and reduced adhesion [51,52], and these effects were associated with the activation of m-calpain (a Ca2+-dependent protease) by reactive oxygen species (ROS) and with the subsequent activations of p38 MAPK and c-Jun N-terminal Kinase (JNK) [51]. In contrast, TRPM7 knockdown by RNA interference increased cell adhesion [52]. TRPM7 is also involved in cell migration. Knockdown of TRPM7 by RNA interference decreased the numbers of high-calcium microdomains induced by platelet-deprived growth factor (PDGF) and inhibited turning by migrating fibroblasts [53]. In cholinergic synaptic vesicles, TRPM7 was found to be involved in transmitter release [54]. In sympathetic neurons, TRPM7 is localized in synaptic vesicles, forms complexes with synaptic vesicular synapsin 1 and synaptotagmin 1, and then interacts with synaptic vesicular snapin [54]. Furthermore, changes in vesicular TRPM7 channel activities were found to alter acetylcholine release [54].
In a smooth muscle cell line (A7r5) derived from rat aorta, exposure to fluid flow rapidly increased endogenous TRPM7 expression and enhanced constitutive NSCC conductance [55]. The rapid activation of TRPM7 by flow reflects the rapid cell membrane fusion of TRPM7-containing vesicles. Furthermore, the rapid vesicular incorporation of TRPM7 protein occurred independently of its kinase domain. It was suggested this flow dependent regulation of TRPM7 expression becomes significant when VSMCs are directly exposed to blood flow resulting from endothelial damage, such as, that caused by atherosclerosis or vascular injury [56].
TRPM7 plays an important role in cardiac biology. Although dispensable in adult ventricular myocardium under basal conditions, TRPM7 is critical for myocardial proliferation during early cardiogenesis [57]. Furthermore, TRPM7 indirectly influences diastolic membrane depolarization and automaticity in sinoatrial nodes by regulating Hcn4 expression [58].
In the gastrointestinal (GI) tract, intestinal pacemaking is the responsibility of interstitial cells of Cajal (ICCs), and TRPM7 protein is an essential molecular component of the NSCCs of ICCs. Thus, TRPM7 protein is a potential target for the pharmacological treatment of motor disorders of the gut [59].
TRPM7 AND DISEASE
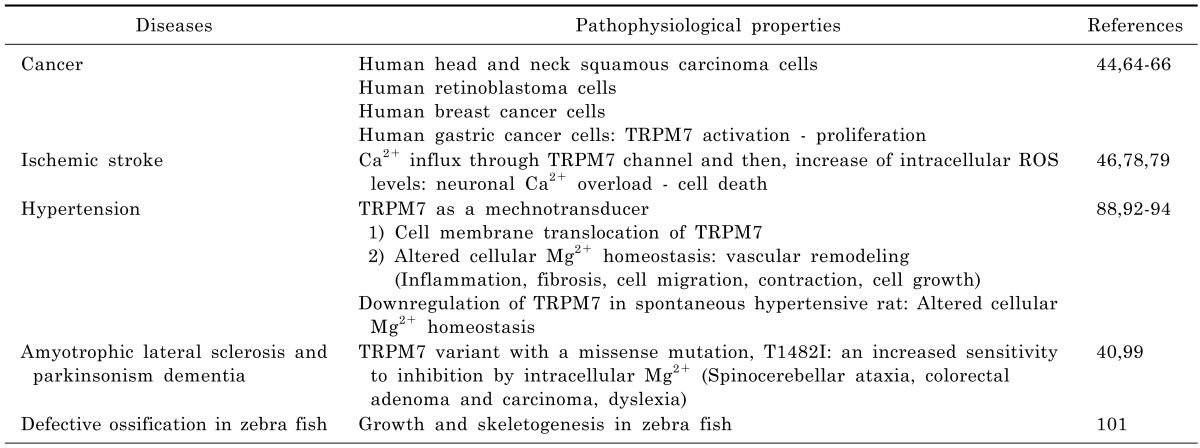
TRPM7 in cancer
Recent years have witnessed increased research interest in the study of the connection between TRP channels and cancer, and this has led to the discovery of tumor-related functions, such as, the regulation of tumor proliferation, differentiation, apoptosis, angiogenesis, migration, and invasion. The TRPC, TRPM, and TRPV family members are believed to be mainly involved in malignant growth and progression. The expression of TRPC6 protein in prostate cancer and the association between it and stage, grade and the androgen responsiveness of tumors have been evaluated [60]. In the TRPV family, the expressions of different TRPV channels (TRPV1, TRPV2, and TRPV6) during cancer growth and progression have been recently described [61]. High levels of TRPM1 expression were found in benign tissues, but its expression was diminished in primary melanoma [62]. TRPM8 mRNAs were found to be over-expressed in prostate tumors by RT-PCR [63,64].
TRPM7 channel is widely expressed in various tissues, including those of the brain, spleen, kidney, lung, liver, and heart [22,42]. Furthermore, its expression has been recently reported in head and neck carcinoma [64], retinoblastoma [44], breast cancer [13], and in gastric cancer [65]. The activation of TRPM7 channels in head and neck squamous carcinoma cell lines enhanced proliferation [64], whereas TRPM7 knockdown by siRNA or blockade of TRPM7 channel by Gd3+ or 2-APB (the latter of which has broad inhibitory effects on TRP superfamily members, including TRPM7) suppressed proliferation. Furthermore, the proliferation of human retinoblastoma cells was promoted by increasing spontaneous Ca2+ influx (achieved by elevating extracellular Ca2+ concentration) [44], and blockade of cation channels by Gd3+, La3+, LOE908, or 2-APB inhibited spontaneous Ca2+ influx and decreased the proliferation of retinoblastoma cells. In addition, it has been shown that TRPM7 knockdown by siRNA transfection significantly reduces Ca2+ influx and retards cell proliferation by delaying G1/S cell cycle progression [44]. TRPM7 channel-related proliferation has also been observed in MCF-7 human breast cancer cells [66] and AGS human gastric adenocarcinoma cells [65], as the proliferations of both were prevented by siRNA transfection induced TRPM7 knockout and by blockading TRPM7 channels with La3+ or 2-APB. TRPM7 channels were found in human healthy breast tissues, and the results were overexpressed in grade III breast cancer in a Ki67 proliferation marker and tumor size dependent manner [66]. TRPM7 was also found to be involved in breast cancer cell metastasis [65]. Thus, TRP channels play important roles as diagnostic and/or prognostic markers, and are targets for pharmaceutical intervention. Accordingly, further investigations are required to improve our understanding of the roles TRP channels in cancer [64].
TRPM7 in ischemic stroke
Cell death during ischemia is often caused by increases in intracellular Ca2+ levels caused by massive glutamate release [67-72]. Excessive Ca2+ entry during ischemia causes irreversible damage to mitochondria, the cytoskeleton, and protein synthesis, and leads to cell death. Therefore, it is important that means of inhibiting excessive Ca2+ influx and release from internal Ca2+ stores are devised. Previously, Ca2+-permeable N-methyl-D-aspartic acid (NMDA) receptors, DL-alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptors [72], and L-type voltage-dependent Ca2+ channels [73] were regarded as the major pathways of Ca2+ entry during ischemia. Thus, blocking of these receptors and channels was considered a potential means of preventing intracellular Ca2+ overload. However, during clinical trials, anti-excitotoxic drugs were found to be ineffective and to have adverse effects. For example, the NMDA antagonist selfotel was administered to patients with ischemic stroke, but was not effective and tended to increase mortality, which suggested neurotoxic effects [74-77]. Therefore, researchers have been examining additional mechanisms of neuronal death, such as, the targeting of non-glutamate-related channels, including TRP channels, to induce ionic imbalance and cell death [78,79].
Interestingly, recent studies have demonstrated the pathophysiological involvement of TRPM7 in stroke. When cortical neurons were subjected to prolonged oxygen glucose deprivation (OGD), intracellular reactive oxygen/nitrogen species (ROS) levels and Ca2+ influx through TRPM7 channel were increased and caused cell death due to a neuronal Ca2+ overload [46]. However, when cortical neurons were transfected with TRPM7 siRNA, ROS-mediated activation and subsequent cell death were reduced. In addition, TRPM7 activated by OGD was found to contribute to ischemia-induced membrane depolarization, intracellular Ca2+ influx, and to the swelling of CA1 hippocampal neurons in brain slices taken during the acute stage [78]. In addition, it is also possible opening of TRPM7 channels induces membrane depolarizations due to Ca2+ influx and the removal of Mg2+ blocking NMDA receptors, thus exacerbating Ca2+ toxicity. The suppression of TRPM7 in the CA1 neurons of adult rats by injecting viral vectors containing a small hairpin RNA (shRNA) specific for TRPM7 resulted in no adverse effects on survival, neuronal or dendritic morphology, neuronal excitability, or synaptic plasticity [79]. Furthermore, TRPM7 suppression protected neuron from ischemic death after 15-min of transient global ischemia, and preserved neuronal function and morphology. These results demonstrate TRPM7 contributes to ischemic neuronal damage, and suggest that TRPM7 be viewed as a potential target for neuroprotective treatment.
TRPM7 in hypertension
Hypertension is one of the most common chronic diseases and a major risk factor of mortality and morbidity [80]. The etiology of primary (essential) hypertension is unknown but it is the most common form of the disease. However, several risk factors have been identified, including genetic, renal, metabolic, race, and environmental factors. More specifically, alterations in the intracellular levels of cations, such as, Na+, Ca2+, Mg2+, and K+, are also risk factors of hypertension [81-85].
Mg2+ is critically associated with the regulation of VSMC function [43], and elevated intracellular Mg2+ levels induce vasodilation and attenuate agonist-induced vasoconstriction, whereas depressed intracellular Mg2+ levels lead to hypercontractility and impaired vasorelaxation. Several studies have suggested that intracellular Mg2+ deficiency causes vascular dysfunction in hypertension, and that normalized intracellular Mg2+ levels improve vascular tone and protect against the development of hypertension [86,87].
TRPM7 has been implicated in a number of vascular pathologies, including hypertension. Angiotensin II, bradykinin (BK), and endothelin-1 regulate vascular TRPM7 acutely by inducing phosphorylation and chronically by increasing TRPM7 expression at the mRNA and protein levels [43,88-91]. Fluid flow and shear stress stimulate TRPM7 by cytosol-to-membrane translocation in VSMCs and increase the amplitude of a native TRPM7-like current [92-94]. These observations suggest TRPM7 might also act as a mechanotransducer, and that altered cellular Mg2+ homeostasis and abnormal VSMC function in hypertension might be related to defective TRPM7 expression/activity, which could in turn contribute to proliferation, inflammation, fibrosis, and contraction, which are important regulators of the vascular remodeling associated with high blood pressure.
VSMCs from Wistar Kyoto (WKY) and spontaneously hypertensive rats (SHR) express TRPM6 and TRPM7. Basal TRPM6 expression is similar in the VSMCs of both, but basal TRPM7 expression is lower in the VSMCs of SHRs. Furthermore, this overexpression of TRPM7 was found to be associated with a significant reduction in [Mg2+]i in SHR cells. In addition, the downregulation of TRPM7, but not of TRPM6, might play a role in the altered Mg2+ homeostasis observed in SHR VSMCs [88]. It was also reported that after exposed to shear stress, numbers of TRPM7 channels near the plasma membrane doubled in VSMCs [79]. These findings show TRPM7 is a mechanosensitive channel and that its activation induces stretch or mechanical sensing in VSMCs.
Several other TRPs, such as, TRPM4, TRPC1, and TRPC6, have been implicated in blood pressure regulation [95,96]. TRPM4 deficient mice display increased sympathetic tone and blood pressure, and increases in plasma catecholamine could explain increased mean arterial blood pressure via its well-known effect on vessel tone and cardiac output [97]. In another study, Trpc6-/- deficient mice exhibited a basal mean arterial blood pressure elevation of ~7 mmHg [95]. This phenomenon could be explained by the compensatory overexpression of TRPC3, which has been reported in Trpc6-/- mice [95]. On the other hand, TRPC3 is a constitutively active non-selective cation channel, and its overexpression leads to enhanced basal and agonist-induced Ca2+ entry into VSMCs, and eventually leads to enhanced contractility [95,98].
TRPM7 in amyotrophic lateral sclerosis and Parkinsonism dementia
Guamanian amyotrophic lateral sclerosis (ALS-G) and Parkinsonism dementia (PD-G) are epidemiologically linked to an environment severely deficient in Ca2+ and Mg2+. These diseases are characterized by progressive motor neuron degeneration, and are among the most common adult onset neurodegenerative diseases [99]. TRPM7 variant with the T1482I missense mutation has been identified in ALS-G and PD-G patients. recombinant T1482I TRPM7 exhibited the same kinase catalytic activity as wild type TRPM7, but heterologously expressed T1482I TRPM7 produced functional channels that show increased sensitivity to inhibition by intracellular Mg2+ [40]. Because the incidence of ALS-G and PD-G has been associated with prolonged exposure to an environment severely deficient in Ca2+ and Mg2+, this variant TRPM7 allele confers a susceptible genotype in such environments. The overall effect of low Mg2+ is to create conditions of high oxidative stress, which is being increasingly recognized as a key player in the initiation and exacerbation of diseases, such as, ALS-PD, and is known to be associated with spinocerebellar ataxia, colorectal adenoma and carcinoma, and dyslexia [40]. However, in a recent study, Hara et al. [100] suggested that TRPM7 is not associated with ALS/PD in the Kii peninsula of Japan, which suggests familial ALS/PD is not caused by the mutation of a single gene, but that it is a complex disease involving genetic and environmental factors. Therefore, further studies are required to deduce the nature of the relationship between ALS/PD and TRPM7 channels.
Defective ossification in Zebra fish
Skeletal development depends on the differentiation and morphogenesis of multiple cell types to generate elements with distinct forms and functions throughout the body. It has been established TRPM7 is required for growth and skeletogenesis in zebrafish, and furthermore, TRPM7 is believed to have potential use in forward genetic approaches designed to reveal the physiological mechanisms responsible for the development of the adult form [101].
CONCLUSION
Recently, many researchers have investigated the physiology, distribution, and functional properties of TRPM7 channel. TRPM7 plays critical roles in cell survival, proliferation, and development. Furthermore, the pathophysiological significance of TRPM7 impacts a broad range of biomedical research, and diseases, such as, cancer, stroke, hypertension, and neurological and cardiovascular diseases, have been shown to be associated with the abnormal function or overexpression of TRPM7 (Fig. 2, Table 1). In some cancers, TRPM7 is highly expressed, and its inactivation or downregulation has been shown to reduce cancer cell viability by inhibiting Ca2+ influx. On the other hand, in ischemic stroke, TRPM7 suppression has been found to protect against ischemic cell death and to preserve neuronal function. In addition, TRPM7 impairment has been shown to contribute to Mg2+ dysregulation and the vascular dysfunction related to hypertension, and inadequate Mg2+ intake and hypomagnesemia could contribute to the pathophysiologies of chronic diseases, such as, hypertension. Mg2+ normally regulates vascular tone and reactivity by modulating Na+, K+, and Ca2+ and by influencing the activities of multiple enzymes. TRPM7 has also been identified to be a unique Mg2+ transporter and a regulator of Mg2+ influx. Much remains to be discovered regarding the functions and regulations of Mg2+ and TRPM7 in health and disease. Finally, we suggest that the significance and function of the TRPM7 kinase domain in the regulation of TRPM7 channels be investigated in detail. We look forward to the deciphering of the common signaling pathways underlying the diverse, specific functions of TRPM7 in different tissues and organs, and to the development of novel pharmacological tools that target TRPM7 in vivo.

 XML Download
XML Download