

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
In neurons of the mammalian central nervous system (CNS), the unique and crucial role of the soma is to determine the output with given synaptic inputs under various conditions. Therefore, the excitability of somatic membranes is a basic index for understanding neuronal functions in various tasks associated with the integrative and plastic modification of information in the brain or pathogenic conditions such as epileptic seizure. Previous studies, including the paper suggesting the original description of synaptic plasticity, have reported that the induction of long-term potentiation (LTP) of synaptic strength is accompanied by changes of intrinsic excitability (IE) of the soma [1,2]. This indicates the involvement of concurrent changes of voltage-dependent ion channel activities in the soma membrane. With the reliance on changes in the expression and/or gating kinetics of voltage-dependent ion channels in the soma membrane, activity-dependent alterations of IE correlated with synaptic plasticity have been observed in various kinds of animals [3]. However, it is still curious if and how synaptic activities mediate the changes of somatic excitability via regulating voltage-dependent ion channels. In the present study, we focused on one type of voltage-dependent ion channels, A-type K+ channels (IA channels), to study the regulation of somatic excitability mediated with synaptic activities.
For a decade, subunits of voltage-dependent K+ channels (Kv channels) contributing to A-type K+ currents (IA) in neurons have been focused on by neuroscientists explaining neuronal functions to modulate dendritic signal processing, action potential (AP) propagation, synaptic integration and the filtering of fast synaptic potentials [4,5,6,7,8,9]. In particular, the NMDA receptor-dependent synaptic LTP in hippocampal neurons regulates the distribution of IA channels in spines and dendrites, indicating the existence of dynamic and active functions of these channels in memory mechanisms [10,11,12,13,14,15]. Activity-dependent down- or up-regulation of IA channels relying on Kv4.2 subunits is also critical for somatic excitability. The reduction of somatic IA mediating the enhancement of intrinsic excitability (IE) has been frequently observed after synaptic LTP induction in hippocampal slices and cultured neurons [12,14]. This phenomenon indicates that IA-mediated somatic processings during synaptic LTP may play important roles in determining neuronal excitability correlated with memory mechanisms, because the alteration of IE is concerned with information storage [16]. Although it is a subject still under debate, previous results in a number of papers have provided evidence that NR2A-containing NMDA receptors located in active synaptic sites dominantly contribute to the induction of LTP, while extrasynaptic NR2B-containing receptors are necessary for LTD in mature neurons [17,18]. Therefore, in plastic changes of IE correlated with synaptic plasticity, it is reasonable to hypothesize that the activation of synaptic NMDA receptors may be required for increasing somatic excitability during synaptic LTP. This hypothesis would provide evidence that the regulation of voltage-dependent ion channels to change IE may be dependent on the type or localization of NMDA receptors.
In this study, we tested whether the downregulation of somatic IA channels was dependent on synaptic or extrasynaptic NMDA receptors in young hippocampal neurons of rats. In the results, the activation of synaptic NMDA receptors but not extrasynaptic NMDA receptors was absolutely required for the reduction of the somatic IA peak. The dependence of IA channel downregulation on synaptic NMDA receptors was also confirmed in an analysis of gating kinetics showing the hyperpolarizing shift of the inactivation property of IA channels. These results suggest a possibility that the activation of synaptic NMDA receptors under various conditions mediates the enhancement of somatic excitability through the biphasic downregulation of somatic IA channels in young hippocampal neurons, contributing to memory mechanisms as well as pathogenic conditions.
METHODS
Hippocampal primary cultures
Hippocampal primary cultures were prepared from embryonic 20-day old Sprague-Dawley rats. The embryonic rats were removed from deeply anesthetized pregnant rats, then transferred to an ice-cold normal Tyrode solution containing the following (in mM): 140 NaCl, 5.4 KCl, 2.3 MgCl2, 10 HEPES, 5 glucose, pH 7.4 adjusted with NaOH. Isolation of the hippocampi from embryonic rat brains was performed in a chamber containing ice-cold normal Tyrode solution under a microscope in a sterilized environment. Dissected hippocampi were transferred to ice-cold minimal essential medium (MEM) containing Earle's salts and glutamine with 10% fetal bovine serum, 0.45% glucose, 1 mM sodium pyruvate, 25 µM glutamate and antibiotics, and then triturated using 200 µL pipettes. The cells were counted and seeded on 12 mm-diameter glass cover slips (Fisher Scientific) coated with poly-L-lysine (Sigma-Aldrich) at a density of 9×104 cells/mL and maintained at 37℃ in 95% air and 5% CO2. After 7 hours, the whole plating medium was changed to Neurobasal (Sigma-Aldrich) medium containing B-27 (Invitrogen), and half of the medium was changed once at DIV 4. All experiments and procedures with animals were performed with the permission from the Animal Care and Use Committee of Jeju National University.
Electrophysiology and data analysis
For electrophysiological recordings, primary dissociated culture neurons of 6-9 DIV were used. Coverslips containing these young hippocampal neurons were transferred to a recording chamber with a continuous flow of recording solution containing the following (in mM) : 145 NaCl, 5 KCl, 2 CaCl2, 1.3 MgCl2, 10 HEPES, 10 glucose, pH 7.4 with NaOH, and bubbled with 95% O2 and 5% CO2. TTX (0.5 µM) was added to the recording solution to block the voltage-dependent Na+ channels. For recording transient K+ currents (i.e. IA), thick-walled, filamented patch electrodes showing a tip resistance of 4~6 MΩ were used. The patch pipettes were filled with an internal solution containing the following (in mM) : 20 KCl, 125 K+-gluconate, 4 NaCl, 10 HEPES, 0.5 EGTA, 4 ATP, 0.3 tris-GTP, and 10 phosphocreatin, and pH 7.2 was adjusted with KOH. During the recordings, the series resistance varied between 8~30 MΩ, and recordings where the series resistance varied by more than 10% were rejected. No electronic compensation for series resistance was employed, and recordings were filtered at 5 kHz in all experiments. Transient and sustained K+ currents were digitally separated using a prepulse protocol after subtracting leak currents. Peak currents were measured at +60 mV after a 200 ms prepulse to either -120 mV or -20 mV. All electrophysiological data were recorded using an Axopatch 200B amplifier (Axon Instruments), and command pulse generation, data acquisition and analysis were performed using Digidata 1322A convertor (Axon Instruments), pClamp 8 (Axon Instruments) and IGOR Pro (Wavemetrics) software. SPSS (SPSS Inc.) and Excel (Microsoft) software were used for further data and statistical analysis. All results in the present study were presented as the mean±SEM, and Student's t-tests were used to examine statistical significance, set to p<0.05 or 0.01.
Drugs treatment
For observing the correlation between somatic IA channels and synaptic NMDA receptors in cultured hippocampal neurons, it was necessary to enhance synaptic transmission in the in-vitro system. Some of the cultured hippocampal neurons were stimulated to increase neuronal activities by adding KCl (20 mM, Sigma-Aldrich) or glutamate (5 µM, Sigma-Aldrich) to culture media for 24 hours or to recording solution for 5 min (for acute responses), and APV (100 µM, Sigma-Aldrich) was added in some cases to confirm the involvement of NMDA receptors. Glycine (200 µM, 5 min) which is well known to induce LTP chemically, was used for selectively activating synaptic NMDA receptors [17,23]. In cases of blocking synaptic NMDA receptors, MK801 (25 µM, Tocris) with KCl (20 mM, for 24 hours) or glycine (200 µM, for one hour) was added to the culture media before recording. Cell viability under each condition according to drug application was confirmed by using MTT assay. Supplemental fig. 1 showed that survival rates of neurons were not reduced by KCl or glutamate overnight treatment, compared with control neurons. More detailed protocols of drug treatments are provided in the results and figure legends.
RESULTS
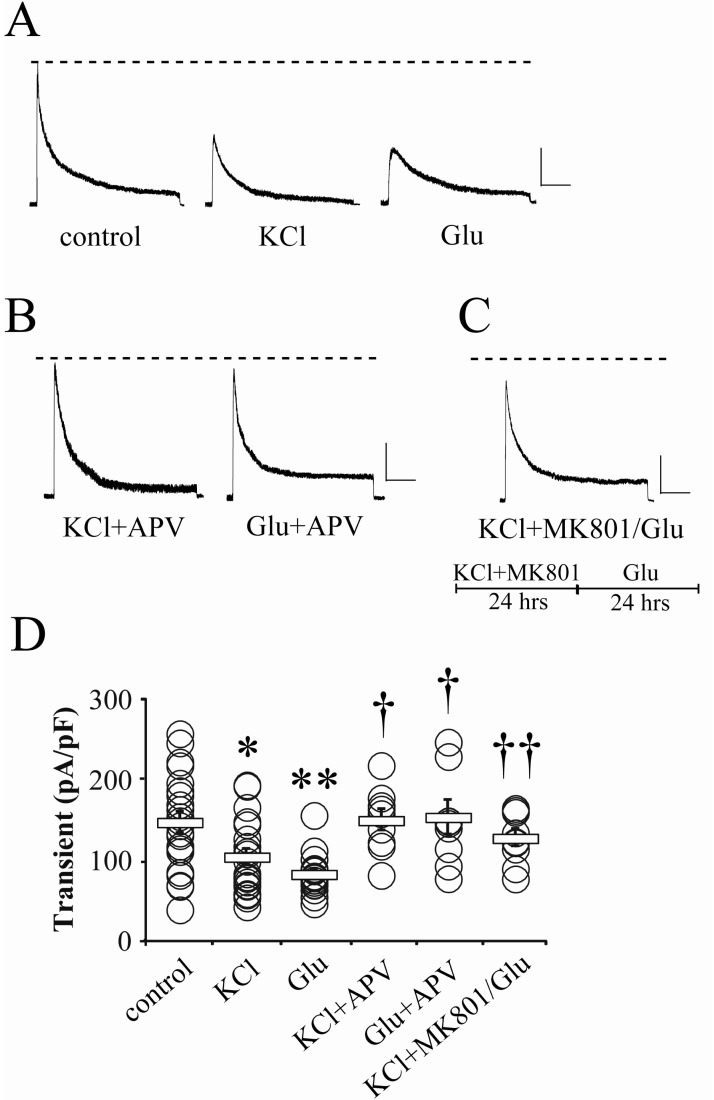
It has been previously reported that the acute activation of glutamatergic receptors induces the internalization of Kv4.2 channels, showing the reduced peak amplitude of somatic IA in electrophysiological results [14]. However, it is not clear whether synaptic or extrasynaptic NMDA receptors contribute to the glutamate-induced reduction of somatic IA peak. In this study, we first focused on the participation of synaptic NMDA receptors in the downregulation of IA channels. All young hippocampal neurons recorded in this study showed invariable whole-cell capacitance within 11~15 pF (n=136). In Fig. 1, we first tested the effect of high KCl on the IA peak recorded by whole-cell patch, because high KCl application can enhance the release of endogenous glutamate from presynaptic terminals, and then activate glutamate receptors in postsynaptic sites [13]. Compared with the control neurons, KCl (20 mM) pretreatment to culture media for 24 hours significantly reduced the peak amplitude of somatic IA (Fig. 1A and D; control=147.49±12.69, n=22; KCl=104.47±9.76 pA/pF, n=21; p<0.05). This indicates that the long-lasting enhancement of glutamatergic transmission may downregulate the somatic IA channels. This KCl effect was mimicked by exogenous glutamate pretreatment (24 hrs, 5 µM; Fig. 1A and D, Glu=82.94±5.98 pA/pF, n=17, p<0.01 compared with the control). Interestingly, both KCl and exogenous glutamate effects were sensitive to APV (100 µM), so the peak amplitude of IA exhibited a level similar with the control group under APV co-application (Fig. 1B and D; KCl+APV=149.87±13.22, n=9; Glu+APV=153.19±21.23 pA/pF, n=9). Because APV is not specific to synaptic NMDA receptors, it seems to be not suitable for testing if synaptic activities are specific for glutamate-reduced somatic IA. In Fig. 1C, we tried to selectively block synaptic NMDA receptors for confirming the specific role of synaptic activities in the reduction of somatic IA. For 24 hours, neurons were cultured in media containing an open channel blocker, MK801 (25 µM) and high KCl (20 mM). This protocol can block irreversibly synaptic NMDA receptors [19]. After washing them out, glutamate was again added to the normal culture media for the next 24 hours. In this experiment, the amplitude of somatic IA was not affected by glutamate application, suggesting a possibility that synaptic NMDA receptors are required for the downregulation of somatic IA channels (Fig. 1C and D; KCl+MK801/Glu=127.75±10.46 pA/pF, n=9, p<0.05 compared with Glu).
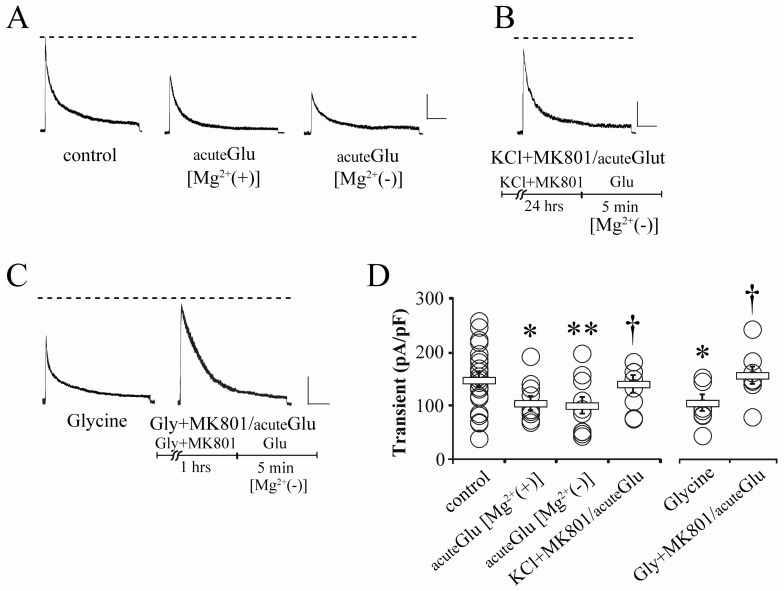
Because Mg2+-sensitive kinetics of NMDA receptors usually lead them to become inactive under the resting condition, MK801-blocked downregulation of somatic IA does not mean the exclusion of extrasynaptic NMDA receptors, Therefore, it is necessary to test if extrasynaptic NMDA receptors can also affect somatic IA regulation. For this issue, we observed the changes of somatic IA in the Mg2+-free recording solution in Fig. 2. Glutamate (5 µM, 5 min) that was acutely added to the recording solution effectively reduced the peak amplitude of somatic IA in both normal and Mg2+-free recording conditions (Fig. 2A and D, acuteGlu [Mg2+(+)]=104.38±13.33, n=10, p<0.05; acuteGlu [Mg2+(-)]=100.04±15.39 pA/pF, n=11, p<0.05; compared with control shown in Fig. 1). However, acute glutamate application did not show any effect on IA amplitude even in Mg2+-free recording solution, when synaptic NMDA receptors were previously blocked by the pretreatment of MK801 (25 µM) and high KCl (20 mM) for 24 hours (Fig. 2B and D; KCl+MK801/acuteGlu[Mg2+(-)]=140.12±16.11 pA/pF, n=10, p<0.05 compared with acuteGlu). This strongly indicates that the downregulation of somatic IA channels is dominantly dependent on synaptic NMDA receptors.
Long exposure to high KCl can damage neurons by inducing overexcitability, and in some cases, resting membrane potentials are significantly depolarized [13]. Although membrane depolarization by high KCl is not sufficient to activate extrasynaptic NMDA receptors, it is necessary to confirm the participation of synaptic activities without significant whole-cell depolarization. In cultured neurons, glycine treatment is useful to enhance synaptic transmission because, as an NMDA receptor coagonist, it activates synaptic NMDA receptors with endogenous glutamate, and consequently, induces chemically synaptic potentiation [14,20]. Glycine (200 µM, 5 min) was added to the recording solution to confirm its effects on somatic IA. In this experiment, glycine significantly reduced the somatic IA peak amplitude (Fig. 2C and D, Glycine=104.76±15.40 pA/pF, n=10, p<0.05 compared with the control). This result is consistent with a previous paper showing the internalization of the Kv4.2 subunit in hippocampal neurons [14]. Because glycine is likely to specifically activate synaptic NMDA receptors in the presence of endogenous glutamate [20], we tried again to block synaptic NMDA receptors by adding glycine and MK801 together. In neurons which were pretreated with glycine (200 µM) and MK801 (25 µM) in culture media for 1 hour before recording, the acute application of glutamate did not reduce the peak amplitude of somatic IA even under Mg2+-free recording conditions (Fig. 2C and D, Gly+MK801/acuteGlu [Mg2+(-)]=156.26±16.15 pA/pF, n=8, p<0.05, compared with acuteGlu [Mg2+(-)]). This clarifies the contribution of synaptic NMDA receptors to the downregulation of somatic IA channels.
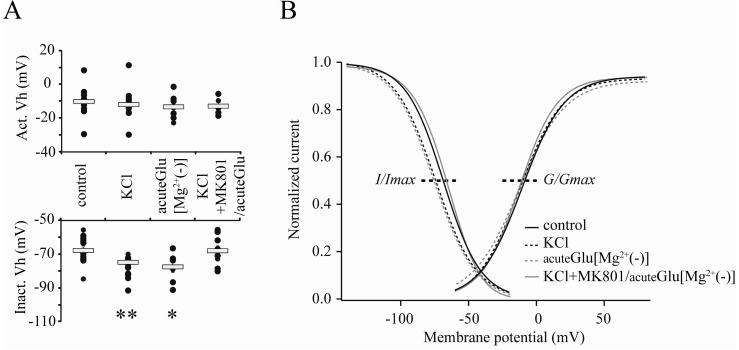
The reduction of IA shown in the present study is supposed to be mediated with the internalization of IA channels from neuronal membranes that are required for the induction of synaptic LTP as well as IE enhancement [12]. In that paper, the hyperpolarizing-shift of the inactivation property of IA channels has been also observed in the initial period of LTP. This change of gating kinetics in IA channel inactivation is strongly correlated with local excitability which is regulated by synaptic activities [11]. In Fig. 3, we also observed the changes of gating kinetics of somatic IA channels. The enhancement of synaptic activities by high KCl (20 mM, 24 hours) or acute glutamate (5 µM, 5 min) induced the hyperpolarizing-shift of the inactivation kinetic of somatic IA channels, while activation properties did not show any significant changes in any of the experiments (Fig. 3; Vhinact; control=-67.69±1.96; KCl=-74.66±2.08, p<0.01; acuteGlu[Mg2+(-)]=-77.40±3.29 mV, p<0.05). This means that, for enhancing neuronal excitability, the gating kinetics of somatic IA channels is also targeted by synaptic activities in hippocampal neurons.
DISCUSSION
The major findings in this study are that the enhancement of neuronal activities significantly reduced the peak of somatic IA in primary hippocampal neurons of DIV 6~9, and that this downregulation of IA channels was specifically sensitive to the activities of synaptic NMDA receptors. Extracellular pretreatment of drugs applied in the present study is not specific to activate especially synaptic receptors in an in-vitro culture system, so it is difficult to simply conclude that enhanced synaptic strength is crucial for influencing somatic IA channels. However, the contribution of synaptic NMDA receptors to somatic IA downregulation has been confirmed by the following observations: 1) that glutamate-induced reduction of somatic IA was completely blocked by the pretreatment of MK801 and high KCl, 2) that the activation of extrasynaptic NMDA receptors in Mg2+-free condition showed no effects on the peak of somatic IA after blocking synaptic NMDA receptors and 3) that the pretreatment with glycine and MK801 abolished the glutamate-induced reduction of somatic IA. Although we did not observe directly synaptic and extrasynaptic components of NMDA currents under each experimental condition in the present study, the pharmacological effectiveness of MK801 and KCl application to isolate extrasynaptic components by blocking synaptic NMDA receptors was previously confirmed by Jung et al. [13]. In that paper, over 50% of total NMDA currents was computed as synaptic component, indicating that synaptic activities are crucial and sufficient to modulate neuronal excitability.
Major functions of IA channels in dendritic processings and synaptic plasticity are dominantly based on the Kv4.2 subunit [13,14]. Because Kv4.2 channels are distributed in dendrites, postsynaptic sites and the soma, the reduction of IA recorded in the soma is possibly due to the internalization of Kv4.2 channels [21,22,23,24]. Also, the somatic regulation of IA channels and their trafficking seems to exhibit similar patterns with those located in dendrites where synaptic plasticity activity-dependently regulates the redistribution of Kv4.2 channels [14]. In cellular mechanisms of memory, enhancement of IE accompanied with synaptic potentiation is a considerable phenomenon to explain memory formation [1,25,26]. The previous paper suggests that the potentiation of somatic excitability (i.e. IE) observed after synaptic LTP induction is dominantly correlated with the reduced IA in somatic membranes [12]. This means that somatic IA channels may be targeted by cellular processings during the induction of synaptic LTP. However, it is still ambiguous if a series of processings for synaptic LTP are required for the regulation of somatic IA channels. In hippocampal neurons, the internalization of dendritic Kv4.2 subunits is sensitive to NMDA receptors [14]. Furthermore, the reduction of somatic IA by glycine suggests the existence of a signaling correlation between somatic IA channel redistribution and glutamatergic transmission, as glycine is an effective agent to induce chemically synaptic LTP in cultured neurons via selectively activating synaptic NMDA receptors [14]. However, it has not been clarified if these somatic and dendritic responses of IA channels are particularly sensitive to synaptic NMDA receptors that are dominant factors to trigger synaptic LTP. In the present study, we blocked synaptic NMDA receptors by pretreating neurons with KCl and MK801 for 24 hours (Fig. 1). KCl treatment is useful to depolarize the membrane as well as to enhance presynaptic glutamate release via activation of the voltage-dependent Ca2+ channels [27]. Therefore, the co-application of MK801 can selectively block the opened NMDA receptors which are located in synaptic sites. The lack of effects of glutamate on somatic IA amplitude in experiments of KCl and MK801 pretreatment indicates that the activation of synaptic NMDA receptors is required for the activity-dependent downregulation of somatic IA channels. This MK801 effect was also observed under the condition of Mg2+-free recording solution to enhance the response of extrasynaptic NMDA receptors, strongly suggesting the specific role of synaptic NMDA receptors.
Recently, it has been demonstrated that the downregulation of IA in cultured hippocampal neurons may be selectively coupled with the activation of NR2B containing receptors located in extrasynaptic sites [28]. In a number of previous papers, NR2B- and NR2A- containing NMDA receptors are dominantly located in extrasynaptic- and synaptic sites in mature neurons, respectively, and their localization and composition pattern determine the direction and degree of synaptic plasticity [17,18]. It is likely that synaptic NR2A-containing NMDA receptors are critical factors for the induction of LTP, while extrasynaptic NR2B receptors trigger the LTD in synapses. However, the reliable correlation of downregulated IA components with the synaptic LTP and enhanced IE suggests the possible involvement of synaptic NMDA receptors [12,13,14]. In the present study, we also observed that the reduction of the somatic IA peak is dominantly mediated by synaptic- but not extrasynaptic receptors. Young neurons of early developmental hippocampi show complicated distribution and composition of NMDA receptors in excitatory synapses. It is also possible that NR2B-containing receptors seem to be still dominantly expressed in synaptic sites of young neurons (~DIV 10), and that the determination of synaptic plasticity is preferentially dependent on the amount of receptors in synaptic sites rather than the type of subunits [13]. Additionally, in Fig. 2, the responses of neurons to acute treatment with glutamate in an Mg2+-free recording solution suggest that the activation of extrasynaptic NMDA receptors is not sufficient for the downregulation of somatic IA in young hippocampal neurons. Our and previous reports indicate that synaptic NMDA receptors may mediate the downregulation of IA channels which are coupled with the enhancement of synaptic strength as well as somatic excitability.
The involvement of synaptic NMDA receptors in the regulation of IA channels has been also reflected in changes of the gating kinetics of IA channels. We also presented the glutamate-induced hyperpolarizing shift of the inactivation kinetics of IA channels in an Mg2+-free solution in Fig. 3, consistent with previous reports showing the NMDA-dependent changes of the gating kinetics of IA channels [11,12]. This shift of inactivation properties of IA channels is important to enhance dendritic excitability and Ca2+ influx and to initiate NMDA-dependent synaptic LTP. However, blocking synaptic NMDA receptors by applying high KCl and MK801 abolished the glutamate-induced shift of IA channels kinetics. These results mean that the activation of synaptic NMDA receptors may mediate biphasic downregulation of IA channels for enhancing neuronal excitability during LTP.
The enhanced efficiency of excitatory postsynaptic potential (EPSP) - AP coupling has been demonstrated as a typical property observed in neurons showing high IE, introducing a possible mechanism for information storage [1,3,12]. Although major cellular changes of synaptic plasticity seem to be restricted within active synapses [29,30,31], other studies even demonstrating the original description of LTP have provided evidence that the induction of synaptic plasticity is accompanied by the alteration of IE in CNS neurons [1,25,26]. This plastic correlation between synaptic and somatic alteration involves potential and concurrent changes of membrane factors such as voltage-gated ion channels to regulate membrane potential of neurons. It has been demonstrated that the enhancement of IE after synaptic LTP induction is clearly mediated through the reduction of the somatic IA peak amplitude in young hippocampal neurons [12]. Furthermore, the somatic and dendritic redistributions of IA channels after LTP induction are not likely to occur independently as both were sensitive to the blockade of clathrin-mediated endocytosis [12,14]. It is necessary to address how synaptic plasticity and associated changes in dendritic processing modulate IE via regulating somatic IA channels in a further study. As the potentiation of synaptic strength increases the local dendritic excitability and subsequent Ca2+ influx [11], it is possible that the expression level of IA channels in a somatic area may then be regulated by neurons for initiating the secondary processing of memory stabilization [16].
In the present study, we summarize that the biphasic downregulation of somatic IA is mediated by the activation of synaptic NMDA receptors in young hippocampal neurons. Although we did not observe cellular linkers between synaptic and somatic processings, the consequential changes of somatic excitability following the activity-dependent modulation of synaptic responses may be a series of processings for neuronal functions to determine outputs in memory mechanisms or pathogenic conditions.
 XML Download
XML Download