

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Parkinson's disease (PD) is a neurodegenerative disorder affecting 1% of population above the age of 65 [1] and is characterized by selective loss of dopaminergic neurons in the substantia nigra pars compacta (SNC). Degradation of the dopaminergic neurons located in the SNC and a subsequent loss of dopaminergic nerve terminals in the striatum are responsible for most of the movement disorders [2]. Although the underlying cause of dopaminergic cell death or process by which these cells degenerate in PD is not clearly understood but oxidative stress [3], mitochondrial dysfunction [4], apoptosis [5,6,7] and protein misfolding [8] are thought to play an important role in causing dopaminergic cell death and cellular degeneration.
Within the central nervous system, tetrahydrobiopterin (BH4) is an essential cofactor for dopamine (DA) and serotonin synthesis. The BH4 is an obligatory cofactor for tyrosine hydroxylase (TH), the first and rate limiting enzyme in the dopamine synthesis and is produced selectively in monoaminergic neurons in the brain including the nigral dopaminergic neurons [9,10,11]. It has been reported that excess DA is potential source of oxidative stress [12] and is major contributing factor for nigrostraiatal degeneration [13,14,15]. It has been demonstrated that BH4 and DA together lead to generation of oxidative stress and render DA cells particularly vulnerable [16] and furthermore administration of BH4 to animals leads to as selective degeneration of the DA system [17]. BH4 itself generates oxidative stress by undergoing auto-oxidation [18,19,20,21] and elicits the formation of reactive quinonoid derivatives of DA and protein as well as lipid peroxidation in DA cells [22]. The amount of BH4 synthesizing enzyme, GTP cyclohydrolase I (GTP-CH I), is lowest, suggesting that the level of BH4 in this area might be normally subtoxic [23,24]. However, it is possible that conditions that lead to aberrant increase in BH4 and DA might elicit oxidative damage at SNC.
It has been reported that transition metal ions like, iron has dopaminergic neurotoxin properties similar to 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) because of its capacity to inducing brain injury through the generation of reactive oxygen species and the acceleration of DA auto-oxidation [25]. The iron related striatal DA depletion or turnover via DA auto-oxidation or oxidative stress on PD animal models have been already reported [26] however the iron related striatal BH4 depletion or turnover via oxidative stress has not been studied yet. Hence, the present study aims to investigate the effect of intranigrial infusion of ferrous citrate on DA and BH4 turnover in rat nigrostriatal system in the progress of PD. We also analyzed GTP-CH I mRNA expression in rat nigrostriatal system in parallel.
METHODS
Animal handing and care
All the experimental protocols involving animals were reviewed and approved by the institutional animal care and use committee (IACUC) of Dankook University. Male Sprague Dawley rats (250~300 g) were purchased from Daehan Biolink, Korea and upon the arrival, the animals were housed three per cage for two weeks at room temperature of 22±2℃ and relative humidity 60~70% on a 12-h light-dark cycle with adequate supply of standard rodent food and water ad libitum.
Unilateral destruction of nigrostriatal dopaminergic neuron
Ferrous citrate was diluted in sterile 0.9% NaCl solution to produce the concentration of 2 mM. The solution was filtered through 0.2 µm Minisart® filter unit and was used for intranigrial administration. On the day of surgery, rats were anaesthetized with intraperitoneally (i.p.) injection of tiletamine hydrochloride (125 mg/kg) and zolazepam hydrochloride (125 mg/kg) and positioned on stereotaxic instrument (Kopf Instruments, Tujunga, CA, USA). The 4.0 µL of ferrous citrate solution (2 mM) was infused through microdialysis probe with glued outlet and tear off membrane (CMA12®, CMA/Microdialysis AB, Stockholm, Sweden) at the flow rate of 0.8 µL/min to the right SNC using stereotaxic coordinates; AP: 3.2 mm, L: 2.1 mm and DV: 2.0 mm relative to bregma by the mouse brain atlas [27]. The injections were delivered over the period of 5 min and probes were kept in situ for an additional 5 min after each injection to avoid the reflux along the injection tract. The sham controls were injected with 4.0 µL of 0.9% NaCl. After surgery, animals were decapitated at different time intervals 0 h, 12 h, 24 h, 48 h, 96 h and 1 wk (n=6) using guillotine cutter and brain was quickly removed. The brain was dissected on an ice plate to separate right (ipsilateral) and left (contralateral) striatum. Both striatum were safely stored at -70℃ for biochemical analysis. Striatum was weighted and homogenized in acetonitrile (1:10, w/w) with 5 µL of IS (250 ng/mL). After homogenizing thoroughly, the DA and BH4 from striatum were further extracted by sonication for 60 sec. The homogenates of brain tissues were centrifuged by 12,000 rpm for 10 min at 4℃. Supernatants were transferred to 96-well plate and then injected onto the LC-MS/MS system by auto-sampler for subsequent analysis.
LC-MS/ MS determination of DA and BH4
The modified method was used for simultaneous determination of DA and BH4 in brain sample by using liquid chromatography with triple quadrupole tandem mass spectrometer (LC-MS/MS) [28]. Briefly striatum was accurately weighed and mixed with acetronitrile containing 200 µg/ml dithiothreitol and 2 µg/ml cimetidine (internal standard). The homogenized mixture was vortexed for 30 sec and centrifuged (8,000 rpm, 10 min) for extraction of DA and BH4 from striatum. The clear supernatant was collected in 100 µL of microtube and was dried under gentle stream of nitrogen at 45℃. The dried extract was reconstituted with 100 µL of 50% methanol containing 0.1% formic acid and 10 µL was injected into LC-MS/MS system. The sample was eluted with mobile phase and was analyzed under the multiple reaction monitoring (MRM) mode: DA (m/z 154 → 177, CE=10 eV), BH4 (m/z 242.2 → 166.0, CE=17 eV), cimetidine (m/z 253.1 → 159.0, CE=14 eV). The liquid chromatographic system was a Accela (Thermo Fisher Scientific Inc., Waltham, MA, USA) system equipped with a solvent delivery module, Nanospace SI-2 3133 (Shiseido Inc., Japan) with an autosampler, and connected to a Discovery Max (Thermo Fisher Scientific Inc.) quadrupole tandem mass spectrometer equipped with electrospray ionization. System control and data analyses were carried out with Xcalibur (Thermo Fisher Scientific Inc.). Chromatographic separation was achieved using a hydrophilic interaction chromatography HPLC column (Luna 3u HILIC 200A, Phenomenex, Torrance, CA, USA) protected by a guard column (Phenomenex C18, 4×2 mm, Phenomenex). A column oven (Nanospace SI-2 3004, Shiseido, Japan) was used online. The mobile phase of 0.1% formic acid-acetonitrile/water (75:25, v/v) was run at a flow rate of 300 µL/min. The electrospray ionization (ESI) mass spectrometer was operated in the positive ion mode.
Total RNA isolation and reverse transcription
The brain tissues were homogenized in TRIzol® reagent (Invitrogen). Total RNA was extracted from the cells according to the manufacturer's suggested protocol. Total RNA (2 µg) was treated with 1 U DNase I (Promega) for 15 min at RT in 18 µL of volume containing 1X PCR buffer and MgCl2 (2 mM). Then it was inactivated by incubation with 2 µL of EDTA (25 mM) at 65℃ for 15 min. Reverse transcriptase reactions were carried out using MuLV Reverse Transcriptase (Promega) according to the manufacturer's protocol. Each reaction tube contained 2 µg of total in a volume of 25 µL containing 5 µL of MuLV 5X RT buffer, 1 µg of oligo-d(T) (promega), 2.5 µL of dNTP Mixture (Promega), 40 U of RNasin (Promega), 20 U of MuLV Reverse Transcriptase and nuclease-free water to volume. Reverse transcriptase reactions were carried out in a DNA Thermal Cycler 480 (Perkin Elmer, Branchburg, NJ, U.S.A.) at 25℃ for 20 min, 42℃ for 60 min and 95℃ for 10 min. The cDNA was then stored at -20℃ until use.
Quantitative real time PCR
Real-time PCR for the analysis of GTP-CH I mRNA levels were performed in a Rotor-Gene 3000 (Corbett life sciences, RG-3000, USA). QuantiTect SYBR Green PCR kit and primers (rat GTP-CH I and GAPDH genes) were purchased from Qiagen. The reaction mixture consisted of 2 µL of cDNA template, 10 µL of SYBR Green PCR master mix and 10 pmol of primers in total volume of 20 µL. The cDNA was denatured at 95℃ for 5 min followed by 40 cycles of PCR at 95℃ for 5 sec, 55℃ for 10 sec and 72℃ for 15 sec. Data acquisition and analysis of real-time PCR were performed using the Rotor-Gene 3000 series software. Deltadelta Ct method was used to calculate the relative quantitation of GTP-CH I normalized with GAPDH level in each individual sample.
Statistical Analysis
All values shown in the figures are expressed as the mean±SEM obtained from at least three independent experiments. Statistical analysis was carried out one-way analysis of variance (ANOVA) with Tukey's post-hoc test (Fig. 2,Fig. 3,Fig. 4) using GraphPad Prism (GraphPad Software, San Diego, CA, USA). Statistically significant differences between groups were assumed when p<0.05.
RESULTS
Histological verification of unilateral intranigral iron infusion
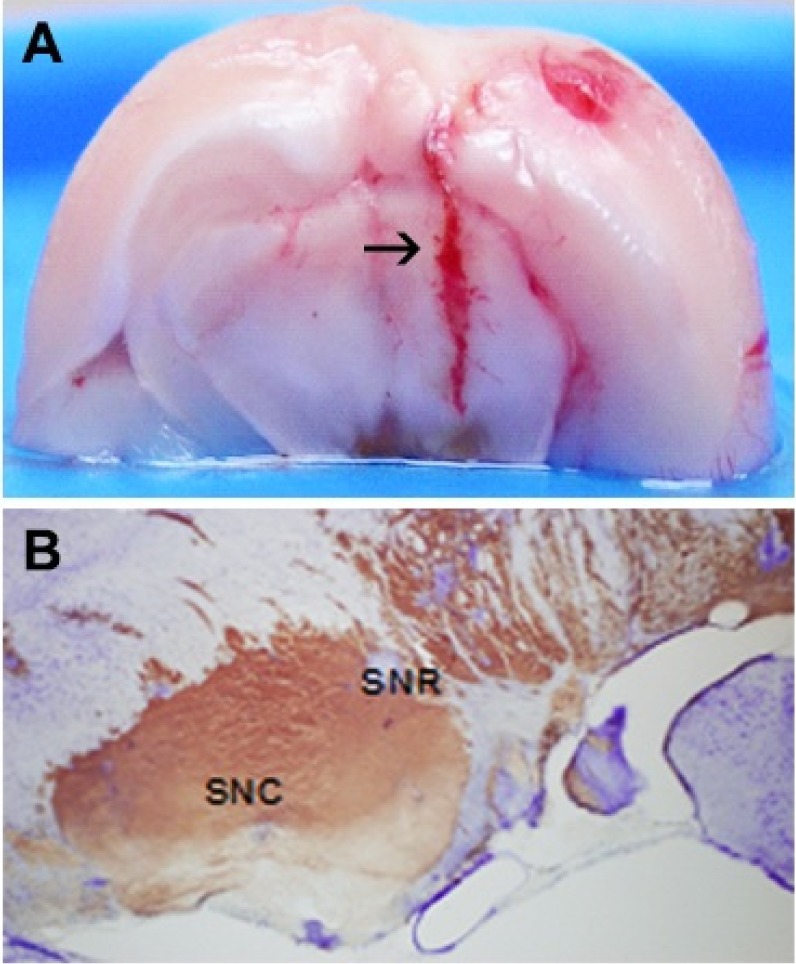
As the preliminary experiment, upon completion of the microinfusion of ferrous citrate into SN, the animals were anaesthetized with i.p. injection of tiletamine hydrochloride (125 mg/kg) and zolazepam hydrochloride (125 mg/kg) and transcardially perfused with 10% formalin (neutral buffered, pH 7.4). The brains were separated and immersed in the fixative for overnight and then brain tissues were cryoprotected after soaking in a series of sucrose solution (10%, 20%, and 30%) at 4℃ until they sank in liquid nitrogen. Serial coronal section, 40 µm were cut with freezing, sliding microtome and stained with cresyl violet. The image shows that injected microdialysis probe tips were located in the SN which was stained with iron (Fig. 1).
Observation of behavioral response after unilateral intranigral iron infusion
After unilateral intranigrial infusion of ferrous citrate, animals showed spontaneous contralateral circling behavior. Although spontaneous circling behavior gradually reduced after lesion, it was exaggerated by the application of painful stimulus. The contralateral circling movement was increased after pinch application by pincette on the left hindlimb or drop of hot water (70℃) by Pasteur pipette on left ear (data not shown). It was suggested that the circling movement after surgery was due to imbalance of nigrostriatal dopamine in lesioned and intact hemispheres, and contralateral circling movement is the key behavioral phenotype of the unilateral lesioned PD rodent model [29]. There were no significance between contralateral circling behavior and DA level (data not shown).
Alteration of striatal DA level
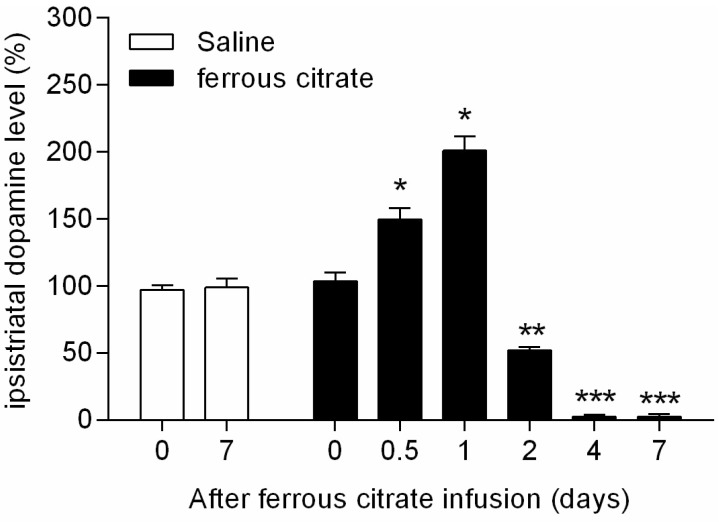
Intranigral infusion of ferrous citrate caused changing DA releases in the striatum via nigrostriatal dopaminergic system. The average concentration of DA in the contralateral striatum (n=12) was 4328.5±26.4 (ng/wet tissue weight g). The percentage ratio of DA concentration in the ipsilateral striatum (n=6) after stereotaxic iron infusion at different time intervals (0, 0.5, 1, 2, 4, and 7 days) were measured. The maximum ipsistriatal DA level was 200.9±10.7% at 1 d and ipsistriatal DA was severely depleted to 2.7±1.1% at 4 d after intranigral infusion of 4 µL of 2 mM ferrous citrate (Fig. 2) The DA levels in ipsilateral striatum by the intranigral infusion of 4 µL of 0.9% NaCl (vehicle, n=4) with respect to percentage of contralateral striatum at 0 and 7 d were 97.0±3.7% and 98.9±6.7%, respectively. There were significant differences of ipsistrialtal DA concentration between 0 d (before injection) and 0.5, 1, 2, 4, and 7 days after intranigral ferrous citrate administration (Fig. 2). These data showed that intranigral iron accumulation caused initial increase in DA levels in nigrostriatal system which induced oxidative stress, preceded chronic decrease in the striatal DA levels such as the progress of PD.
Alteration of striatal BH4 level
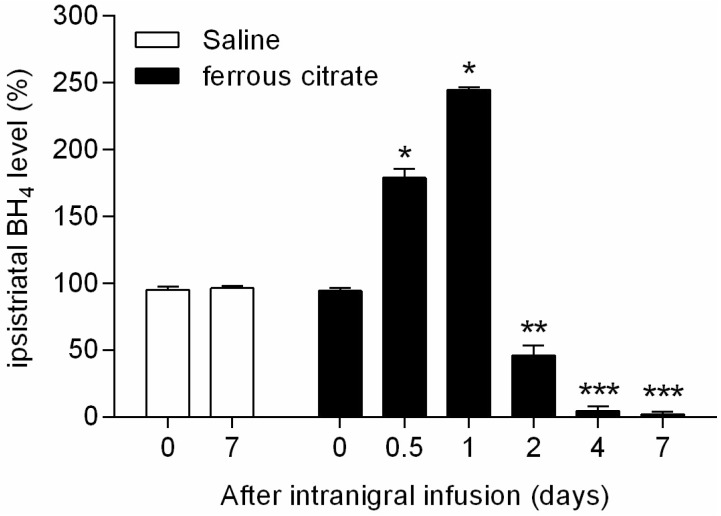
Intranigral infusion of ferrous citrate caused changing BH4 release in the striatum by nigrostriatal dopaminergic system in a similar pattern of DA release. The average concentration of BH4 in the contralateral striatum (n=12) was 192.1±19.0 (ng/wet tissue weight g). The percentage ratio of BH4 concentration in the ipsilateral striatum (n=6) after stereotaxic iron infusion at different time intervals from 0 to 7 d were measured. The maximum ipsistriatal BH4 level was 244.7±2.0% at 1 d and ipsistraital BH4 was severely depleted to 4.5±3.5% at 4 d after intranigral iron infusion (Fig. 3) The BH4 levels in ipsilateral striatum by the intranigral infusion of vehicle (n=4) with respect to percentage of contralateral striatum at 0 and 7 d were 94.9±12.3% and 96.3±11.7%, respectively. There were significant differences of ipsistrialtal BH4 level between 0 d (before injection) and 0.5, 1, 2, 4, and 7 days after intranigral ferrous citrate administration (Fig. 3). These data showed that intranigral iron accumulation caused initial increase in BH4 levels in nigrostriatal dopaminergic neurons, which preceded chronic decrease in the striatal BH4 levels. The result showed that the turnover of BH4 and DA were significantly correlated (r=0.968, p<0.05) after iron infusion. However, striatal BH4 concentration increased further than DA concentration until 1 d but interestingly, decreased quickly after 1 d compared with striatal DA turnover in rat nigrostriatal system. It is suggested that the BH4 concentration changes more dynamically than the DA concentration in striatal system (Fig. 2 and 3).
Changes of GTP-CH I mRNA expression
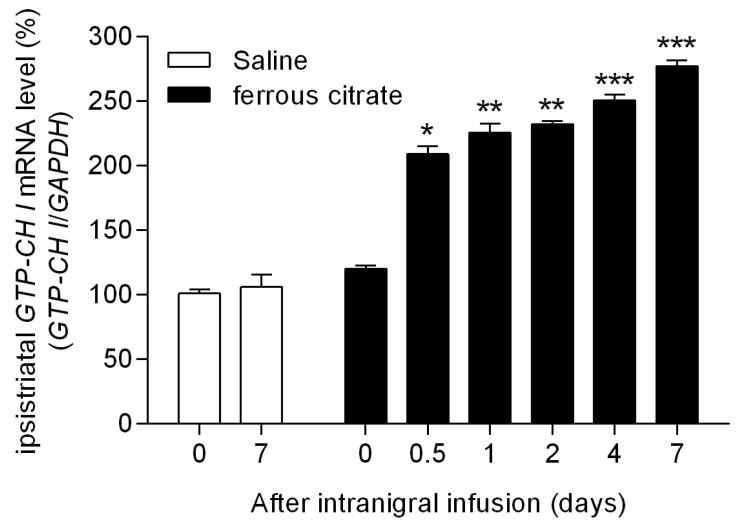
Intranigral infusion of ferrous citrate induced persistent increase of GTP-CH I mRNA expression in the striatum. The percentage ratio of quantitative real time PCR values for GTP-CH I mRNA expression on the ipsilateral striatum (injected side, n=6) to the contralateral side after stereotaxic iron infusion at different time intervals from 0 to 7 d were increased gradually up to 277.4±4.4% at 7 d after ferrous citrate administration (Fig. 4). The GTP-CH I mRNA expression by the intranigral infusion of vehicle with respect to percentage of ipsi-/contra-lateral striatum at 0 and 7 d were 101.2±3.2% and 106.2±9.3%, respectively. There were significant differences of GTP-CH I mRNA expression between 0 d and 0.5, 1, 2, 4, and 7 days after intranigral ferrous citrate infusion (Fig. 4). These data showed that intranigral iron accumulation induced GTP-CH I mRNA expression immediately and the induction of gene expression was sustained at least 7 d in striatum, suggesting increased GTP-CH I mRNA expression might be a compensatory process of BH4 which depleted by the degenerative progress of nigrostriatal dopaminergic neurons.
DISCUSSION
It has been known that trace metal, iron, was accumulated in brain with aging and induced oxidative stress in the basal ganglia, especially in the A9 dopaminergic neurons of SNC area [29]. Interestingly, non-heme iron was increased further in the SN of patients with PD [30,31,32]. In the present study, we had microinfused ferrous citrate, a small molecular weight iron complex, into the SNC area unilaterally as the pathophysiological animal model mimicking PD to study the pathogenic path of the PD. We found that iron caused a chronic depletion of striatal DA and BH4 levels, which was preceded by initial increase in striatal DA and BH4 concentration. In line with our findings, BH4 levels are decreased at 24 h in MPP+ (1-methyl-4-phenylpyridinium)-induced human dopaminergic model of PD [33]. Previous our findings show that MPTP induce the reduction of DA but not BH4 level in striatum and SN [34]. Intranigral iron infusion induced nigral DA and BH4 release into striatum peaked during 0.5 and 1 d after the stereotaxic procedure which suggested that initial increased synthesis and/or release of nigral DA and BH4 induced by iron accumulation augmented the oxidative damage to nigrostriatal dopaminergic neurons, and damaged nigrostriatal dopaminergic neurons represented reduced synthesizing capacity of DA and BH4 as the biochemical phenotype in PD. Therefore, this intranigral iron infusion is a quite reliable model for PD because it showed both biochemical and behavioral phenotypes of PD with slow progress even similar neurochemical and behavioral changes have been documented following infusion of dopaminergic neurotoxin MPTP and 6-hydroxydopamine in the nigrostriatal dopaminergic system [34,35,36].
It has been previously proposed that oxidative injury in PD may be caused by oxidants generated during enhanced DA turnover or iron-catalyzed DA auto-oxidation [37,38]. Iron induced hydroxyl radical generation and lipid peroxidation may be caused, at least in part, by sustained DA turnover which was observed during first two days after iron infusion. Earlier reports indicated that iron induced striatal DA depletion is linked to the loss of tyrosine hydroxylase- positive neurons in the SN. The present study demonstrates that the degree of the nigrostriatal degeneration is correlated with, not only DA turnover, but also chronic depletion of striatal BH4. Several studies showed that both hydroxyl radicals and iron have been implicated in the initiation and propagation of chain reaction of lipid peroxidation [30,31,32]. It has been well documented that iron caused sustained lipid peroxidation which induced cell membrane damage, calcium overload and eventually lead to cell death when the oxidative stress overwhelms the cellular integrity [31]. Elevated non-heme iron is routinely found in the SNC in PD patients [32]. As suggested by Kim et al. [39], it is possible that an aberrant increase in BH4 and DA might elicit oxidative damage in the SNC, and resulting structural damage to nigrostriatal pathway.
BH4 auto-oxidation could produce hydrogen peroxide and superoxide radical [18,19,20,21]. In addition, DA, with its synthesis facilitated by BH4, in turn undergoes auto-oxidation to form a reactive quinine species [18,22,24]. Indeed, DA derived quione production is elevated in which BH4 provides its auto-oxidation products [18]. As both BH4 and DA are inevitably produced within the same cells for DA neurotransmission, the DA cells would tend to be more vulnerable especially under intranigral iron infusion conditions that result in overproduction of BH4 and DA.
All the monoaminergic neurons and terminals in the CNS produce BH4 and DA cells are vulnerable to high level of BH4, most probably due to the higher rate of oxidation of DA compared with other catecholamine or serotonin [18,19]. Under, the normal conditions, the level of GTP-CH I expression in the nigrostriatal system is lower than in the other monoaminergic cells [23,24]. In addition, DA is normally sequestered in the storage vesicles by the action of vesicular monoamine transporter (VMAT) after synthesis in the cytosol, limiting its encounter with BH4 [23]. Therefore, an aberrant condition such as intranigral iron infusion, would lead to initial overproduction of BH4 and DA as the consequence of oxidative damage and then gradually depress the production of BH4 and DA, in which GTP-CH I levels were increased after 24 h with compared to sham control (vehicle infused group).
This is the first study for demonstrating an overproduction of BH4 and DA actually occur in vivo in PD animal model. Iron accumulation in SN elevated BH4 and its synthesizing enzymes, GTP-CH I and DA. This result provides a strong support for current view in progression of PD that excessive DA is a contributing factor for nigrostriatal degeneration. In addition, we demonstrate that intranigral infusion of iron could generate a PD animal model for studying pathophysiology and for developing preventive method of PD progress in vivo. In the present study we showed iron accumulation in SN causes increases in production of both BH4 and DA and this concomitant elevation induces degeneration of the nigrostriatal pathway. As BH4 can increase oxidative damage by auto-oxidation as well as by enhancing synthesis of DA, whose oxidation is in turn facilitated in the presence of BH4, it is possible that the iron accumulation in the dopaminergic neuron can increase BH4 levels, which might be a primary etiologic factor of PD in aged brain.
 XML Download
XML Download