

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Epothilones, a class of polyketide-derived 16-membered ring macrolides, were initially discovered as the cytotoxic metabolites of a soil myxobacterium Sorangium cellulosum [1]. These macrolides were subsequently identified as microtubule-stabilizing agents; they have the similar mode of action as paclitaxel, but are efficacious to paclitaxel-resistant tumors [2]. In the last decade, there has been much interest in the use of epothilones to develop novel antineoplastic agents with several candidates being under clinical investigations [3]. Ixempra (ixabepilone), as an important milestone, has been approved by FDA as a therapeutic anti-cancer drug to treat metastatic and locally advanced breast cancer patients [4]. UTD2 is an epothilone derivative generated from the genetic manipulation of the epothilone biosynthetic gene cluster. Based on the structure-activity relationship studies, structural changes in the molecule may offer an improved antitumor activity. UTD2 has demonstrated a high level of activity against a broad range of tumors both in vitro and in vivo, which include paclitaxel-sensitive tumors and multi-drug-resistant carcinoma models (Qiu et al., unpublished data).
Previous studies concerning the molecular action of epothilones have mostly focused on the microtubule targeting, including the pathway to mitotic arrest and apoptosis induction [2]. So far, the effects of epothilones on the important signaling pathways for the regulation of cytoskeleton and cell growth/survival have not been reported. Due to the broad range of structural and regulatory interactions between the microtubules and the actin cytoskeleton, the possible effects of novel epothilones on actin cytoskeleton-related cellular processes should be investigated and gain more insight into the relevant signaling pathways. Rho GTPases play pivotal roles in actin cytoskeletal reorganization. The existing data derived from in vivo and in vitro studies also indicate that Rho GTPase signaling pathways regulate the growth, motility, invasion and metastasis of breast cancer cells. Alteration of either the expression or the activation status of different Rho GTPases can lead to different pathologies [5]. We therefore sought to determine whether different Rho GTPases show different sensitivities to epothilones during the treatment of cancer cells and to clarify the downstream signaling pathways of Rho GTPases that may be affected by UTD2 in breast cancer cells. In this study, we focused on the effects of the novel epothilone analog UTD2 on actin cytoskeleton-related cellular processes and on the activities of two critical regulators of the cytoskeletal system, namely, the small Rho GTPases Rac1 and Cdc42. The downstream signaling molecules of Rac1 GTPase important for actin cytoskeletal organization and cell proliferation were also assessed.
METHODS
Chemicals and reagents
Novel epothilone analogs were obtained from Biostar Technologies (Beijing, China). All the cell culture reagents were purchased from Invitrogen (San Diego, USA). MTT, Triton X-100, β-tubulin antibody, Hoechst 33342, and Matrigel were purchased from Sigma-Aldrich (St. Louis, USA). Rac1, Cdc42 and p-PAK1 primary antibodies were obtained from Santa Cruz Biotechnology (Santa Cruz, USA). p-ERK1/2, p-JNK, p-p38, p-Akt, PAK1, ERK1/2, JNK, Akt and p38 antibodies were purchased from Abcam (Cambridge, UK). Horseradish peroxidase (HRP)-conjugated IgG and FITC-labeled mouse IgG were acquired from Jackson ImmunoResearch Laboratories (West Grove, USA). bFGF and fibronectin were obtained from R&D systems (Minneapolis, USA). The ECL regent was purchased from GE Healthcare Life Science (Pittsburgh, USA). The polyvinylidene fluoride (PVDF) membrane was acquired from Millipore (Massachusetts, USA). Superscript II reverse transcriptase was the product of Takara (Dalian, China). UTD2 was initially dissolved in dimethyl sulfoxide (DMSO) as a stock solution and subsequently diluted to the desired experimental concentrations with culture media.
Cell culture
The human breast cancer cell lines MCF-7, MDA-MB-231 were purchased from the American Type Culture Collection (ATCC) (Manassas, USA). The stable cell lines expressing constitutively active Rac1 or dominant negative Rac1 were established by transfecting MCF-7 cells with the myc-tagged Rac1 mutant expression plasmids, that is, myc-tagged Rac1V12 (mRac1V12)and myc-tagged Rac1N17 (mRac1N17), and selected for stable expressing clones. The expression of the myc-tagged Rac1 mutants was screened by Western blot using anti-myc tag antibody. The cells were cultured in Dulbecco's modified Eagle's medium (DMEM), supplemented with 10% fetal calf serum (FCS), 100 U/mL penicillin and 100 µg/mL streptomycin at 37℃ in 5% CO2.
In vitro antiproliferative assay
The antiproliferative effects of novel epothilone analog UTD2 were assessed by 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) assay as described before [6]. The UTD2 stock solution was further diluted to the appropriate concentrations in culture media immediately before use. The cells were plated in culture medium in 96-well plates (3000 cells per well, in quadruplicate) and were treated with UTD2 at concentrations ranged from 0 to 1000 nM. The cells were incubated with the drug for up to 72 hours and the number of viable cells was then determined. The results were expressed in terms of IC50 and IC10 (the highest non-toxic concentration of a drug, HNTC) values, which represent the drug concentrations responsible for 50% and 10% inhibition in the growth of cells, respectively, compared to the control cell growth. Both the IC50 and the IC10 concentrations were used in further experiments.
Immunocytochemistry
Breast cancer cells were seeded on glass coverslips in a 24-well plate. At 80% confluence, the cells were treated with or without UTD2 (IC10, IC50) for 24 hours and then were fixed with 4% Polyoxymethylene. Before fixation, cells were not washed with phosphate-buffered saline (PBS) to preserve their cytoskeletal integrity. The fixed cells were then incubated with 5% bovine serum albumin (BSA) in PBS for 30 min to block non-specific reactions. After blocking with BSA, the cells were permeated with 0.1% Triton X-100 in PBS and incubated with β-tubulin antibody (1:50) for 2 hours. After three PBS washes for 15 min, the cells were incubated with FITC-labeled anti-mouse IgG (1:120) for 30 min to visualize β-tubulin. After PBS washes, the cells were incubated with Hoechst 33342 (10 µg/mL) for 10 min to stain the nuclei. The images were captured by confocal laserscan microscope through a 100×objective lens and subsequently analyzed with SlidebookTM software (Version 4.0; Intelligent Imaging Innovations Inc., USA).
Monolayer wound healing assay
MCF-7 cells were seeded in a 6-well plate at a density of 1.5×106 cells per well. Before plating, two parallel lines were drawn at the underside of each well with a marker. These lines will serve as fiducial marks for the wound areas to be analyzed. The cells were serum starved overnight before performing the assay. On the day of analysis, the monolayer was completely confluent. Two parallel scratches that were approximately 200 µm in width were made perpendicular to the marker lines with a sterile pipette tip. This procedure allows the entire width of the wound to be imaged using a 10× objective lens. The wounds were observed using phase contrast microscopy on an inverted microscope.
Migration assay
The cells migration was measured in a 24-well plate transwell system containing inserts with a fluorescence blocking filter and a pore size of 8 µm. MCF-7 cells were seeded in the insert at 105 cells per well. The cells were treated with UTD2 (IC10, IC50) for 16 hours or culture media as control. The upper media was serum free, culture media containing 20% FCS and 30 ng/mL Basic fibroblast growth factor (bFGF) were added to the lower compartment of the system to elicit tumor cell migration. The cells were allowed to migrate for 16 hours at 37℃ in 5% CO2. Cotton swabs were used to clean off the non-migrated cells on the top of the transwell. To fix the migrated cells, 4% Polyoxymethylene dissolved in PBS was applied for 15 min at room temperature, and 0.1% crystal violet was used to stain the cells for 10 min. The migrated cells are observed using phase contrast microscopy on an inverted microscope.
Invasion assay
The breast cancer cells (MCF-7) invasion was measured in a 24-well plate transwell system containing inserts with a fluorescence blocking filter and a pore size of 8 µm. The inserts were coated on the bottom with 2 mg/mL fibronectin, washed with PBS and coated on the upper side with 5 mg Matrigel in 100 mL medium. A number of 105 tumor cells were seeded on top of the Matrigel layer in each well and were allowed to settle for 16 hours. Meanwhile, cells were exposed to UTD2 (IC10, IC50), or were left untreated (control) followed by replacement by culture media without FCS in the upper compartment. Culture media containing 20% FCS and 30 ng/mL bFGF was added to the lower compartment of the system to elicit tumor cell invasion through the Matrigel layer. 16 hours later, the non-invasive cells on the top of the transwell were clean off by cotton swabs. 4% Polyoxymethylene dissolved in PBS fixed the invasive cells for 15 min at room temperature, and 0.1% crystal violet stained the cells for 10 min. The invasive cells are observed using phase contrast microscopy on an inverted microscope.
Adhesion assay
The assays of cell adhesion to immobilized Matrigel were based on that described in Faull et al. [7]. 200 µL dilutions of 4 mg/mL Matrigel in media without serum were incubated inside the wells of a 96-well plate 30 min at 37℃. After washing with PBS, the wells were blocked with 2% BSA in PBS for 2 hours at room temperature. Cells were washed three times in PBS and resuspended at 106 cells/mL, and 50 µL aliquots were added to each coated well. To test the effect of UTD2 on the cell adhesion, the cells were preincubated with UTD2 (IC10, IC50) for 30 min at room temperature and washed twice, before addition to the wells. After 2 hours incubation at 37℃ and 5% CO2, the non-adherent cells were washed off with two rounds of gentle pipetting, the residual adherent cells were fixed with 4% Polyoxymethylene dissolved in PBS for 15 min at room temperature, and the cells were stained with 0.1% crystal violet for 10 min. Stained cells were washed three times with PBS. After washing, the staining reaction was terminated by the addition of 50 µL of 1% SDS and read in an ELISA plate reader with a 570 nm filter. Background values were determined in wells coated with 2% BSA.
Rac1/Cdc42 activation assay
The breast cancer cells were grown to 80% confluence in 10 cm petri-dishes and rendered quiescent in culture media without FCS overnight. Rac1/Cdc42 activation was initiated by the addition of complete culture media (10% FCS), while 5 nM, 10 nM or 20 nM UTD2 were added concurrently. After four hours, Rac1/Cdc42 activity was analyzed using a Rac1/Cdc42 activation assay. Briefly, after a washing step, the cells were lysed in the assay buffer, which was supplemented with 0.5 mM trypsin inhibitor, 0.5 mg/mL leupeptin, 1 mM PMSF, and subsequently centrifuged to remove cell debris. The cell lysate was incubated with 10 mg of agarose that was conjugated with p21-binding domain of PAK1 which binds both activated (GTP-bound) Rac1 and Cdc42, overnight at 4℃. Agarose beads bound with activated Rac1/Cdc42 were washed four times in the assay buffer, resuspended in 30 µL SDS-sample buffer (10% glycerol, 3% SDS, 20 mg/mL bromophenol blue, 25 mg/mL β-mercaptoethanol, 0.6 M Tris, pH 6.7). The samples were subjected to 12% polyacrylamide gel electrophoresis. The activated Rac1/Cdc42 protein levels in each sample were analyzed by Western blot using a rabbit polyclonal antibody against Rac1 (1:1000), or a mouse monoclonal antibody against Cdc42 (1:1000).
Zymography
The activity of matrix metalloproteins (MMPs) MMP-2 was determined using slightly modified substrate-impregnated gels. The breast cancer cells MDA-MB-231 were plated in a 24-well plate with 3×105 cells per well and then treated with UTD2 (0 nM, 5 nM, 10 nM or 20 nM) for 24 hours. After each treatment, the culture media was replaced with serum-free media and incubated overnight. The culture supernatants were collected and proteins were separated on a gelatin-impregnated (1 mg/mL) 10% SDS-PAGE. The gels were then incubated for 18 hours at 37℃ in 50 mmol/L Tris, 0.2 mol/L NaC1, 5 mmol/L CaC12, and 1% Triton X-100 (pH 7.6) and stained with 0.5% Coomassie blue G 250 in methanol/acetic acid/H2O. The gelatinolysis was visualized as transparent bands.
RNA isolation and RT-PCR analysis
The MDA-MB-231cells were seeded in 6-well plate, and then treated with UTD2 (0 nM, 5 nM, 10 nM or 20 nM) for 24 hours. The total RNA was isolated using Trizol reagent on the treated cells. Aliquots of 1 µg of total RNA were used for the first-strand cDNA synthesis. The reaction volume was 50 µL and used 100 units of Superscript II reverse transcriptase. The primers [8] that were used for human MMP-2 amplification were the following: 5'-GTGCTGAAGGACACACTAAAGAAGA-3' (sense) and 5'-TTGCCATCCTTCCAAAGTTGTAGG-3' (antisense). The primers used for β-actin were the following: 5'-GTGGGGCGCCCCAGGCACCA-3' (sense) and 5'-CCTTAATGTCACGCACG-3' (antisense). The PCR amplifications were run for 30 cycles at 94℃ for 30 seconds, 55℃ for 30 seconds, 72℃ for 1 min, and 72℃ for 7 min; the samples were then held at 4℃. The PCR products were analyzed via electrophoresis in 1% agarose gels and staining with ethidium bromide.
Focus formation assay
The focus formation assay was carried out according to that described by Qiu et al. [9]. NIH 3T3 cells were transfected with plasmid constructs expressing activated Rac1 and Raf, and the transfected cells were incubated in culture media containing UTD2 (IC50) over night. The cells were then maintained in drug-free media. The number of transformed foci induced by Rac1, Raf alone or by Rac1 plus Raf were quantified about 17 days post transfection. Transfections were performed in triplicate, and data represented the average of three independent experiments.
Western blot analysis
Cells were plated in 10 cm petri-dishes with 8×106 cells per dish. The cells were grown to about 80% confluency. The culture media was replaced by media containing the novel epothilone analog UTD2 (0 nM, 5 nM, 10 nM or 20 nM), and the cultures were further incubated for 24 hours. The treated cells were lysated as described in Ahn et al. [10]. Briefly, the treated cells were gently washed three times with cold PBS, scraped into ice-cold lysis buffer (150 mM NaCl, 20 mM MgCl2, 2 mM EGTA, 50 mmol/L Tris pH 8.0, 1%Triton X-100) and lysed for 20 min on ice. The lysates were centrifuged at 12,000 ×g and 4℃ for 10 min. Samples were first separated by 12% SDS-PAGE and then transferred to a PVDF. The membrane was blocked with 5% nonfat milk in PBS for 1 hour, incubated overnight with primary antibodies (either anti-phosphorylated PAK1 (pS199/204), anti-phosphorylated JNK (pT183/Y185), antiphosphorylated ERK1/2 (pT202/Y204), anti-phosphorylated p38 (pT180/Y182), anti-phosphorylated Akt (pS473), anti-GAPDH), and then probed for 1 hour with secondary HRP-conjugated anti-mouse or anti-rabbit IgG. After extensive washing with PBST, the target proteins were detected on the membranes by enhanced ECL reagent.
Statistics
The results are presented as the mean±SE for each treatment group in each experiment. The data were subjected to statistical analysis by one-way ANOVA, which was followed by the Scheffe post-hoc test, which used SPSS software (SPSS Inc., USA). A probability value of p<0.05 was considered statistically significant.
RESULTS
Determination of the highest non-toxic drug concentration (HNTC, IC10) and IC50 of UTD2 in breast cancer cells
The cells were exposed to UTD2 for 72 hours. The antiproliferative effects of the drug on MCF-7 and MDA-MB-231 cells were determined by the MTT assay. The HNTC (IC10) and the IC50 values are shown in Table 1. UTD2 inhibited 50% of the growth of breast cancer cells at 16 nM. To ensure the validity of the IC10 value, the inhibition of cell growth had to be less than 10% as compared to the control cells. The last column of Table 1 gives the mean percentage of cell growth when breast cancer cells were treated with the IC10 concentration of UTD2.
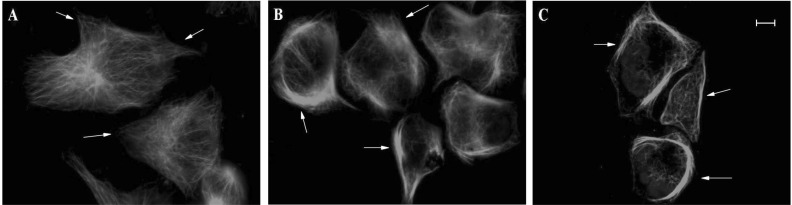
Novel epothilone analog disturbs cytoskeleton organization
The microtubule cytoskeletal changes in the MCF-7 were examined after 24 hours of drug exposure by confocal laserscan microscopy using an anti-tubulin antibody. The untreated MCF-7 cells showed good distribution and integrity of the microtubules throughout the cell, especially in the direction of movement (Fig. 1A arrowhead). The treated cells were more compact. The microtubules were still visible after exposure to UTD2 at IC10, but the filiforms were shorter, and tubulin was primarily present closer to the nucleus (Fig. 1B arrowhead). The MCF-7 cells treated with UTD2 at IC50 showed no clear movement in any direction, only a thick layer of tubulin around the nucleus (Fig. 1C arrowhead). These results indicate that UTD2 treatment led to significantly higher signal intensity of tubulin around the nucleus as compared with that of control cells. Furthermore, exposure to very low concentrations of UTD2 appears to be sufficient to impair microtubule integrity and localization of the tubulin.
Cell movement is coordinated by the actin cytoskeleton [11]. UTD2 treatment seemed to eliminate the movement of MCF-7 cells, suggesting that the drug may act on the actin cytoskeleton. We then investigated the effect of UTD2 on actin cytoskeleton-related cell processes, such as motility, migration, adhesion and invasion, as well as on the activity of the critical modulators of the actin cytoskeletal organization.
The novel epothilone analog inhibited wound closure
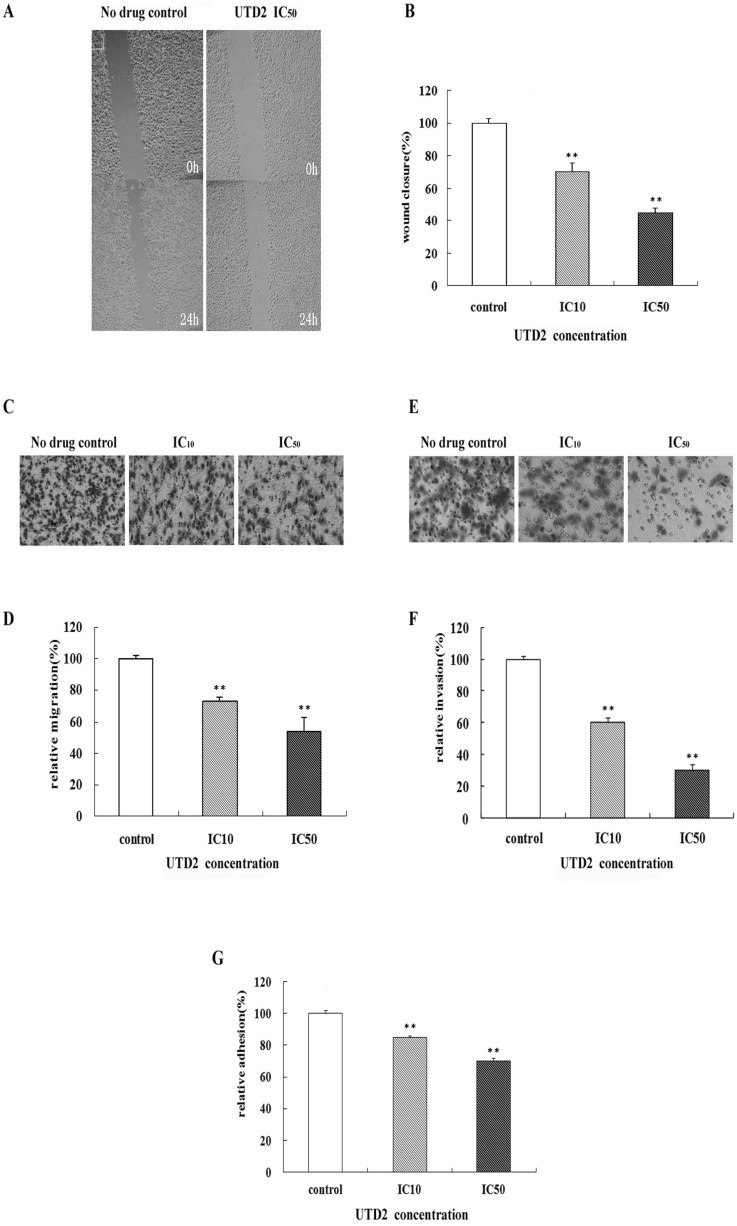
The wound closure was imaged under microscope at different times (up to 24 hours) and was illustrated by a representative experiment (Fig. 2A). The control cells filled the wound by 50% after 24 hours. Treatment with UTD2 significantly inhibited the wound closure abilities of the MCF-7 cells at the IC50. Fig. 2B depicts the wound closure at 24 hours after treatment as a percentage of control wound width. Only 70% of the wound was filled by the cells treated with the IC10 of UTD2 after 24 hours compared to the control wound width. After exposure to the IC50, the wound closure was only 40% of the control width (Fig. 2B, p<0.01).
Novel epothilone analog inhibited migration and invasion of breast cancer cells
The transwell system was used to investigate the effects of UTD2 on migration and invasion. Our results demonstrated that UTD2 inhibited the migration of MCF-7 (Fig. 2C). Compared to the control cells, UTD2 treated cells exhibited only approximately 70% cell migration at the IC10 and 50% at IC50 concentration (Fig. 2D, p<0.01).
Novel epothilone analog inhibited adhesion of breast cancer cells
To study the effect of UTD2 on the ability of the breast cancer cells to attach to the extracellular matrix (ECM), an adhesion assay was performed. The results are displayed in Fig. 2G. Without the addition of UTD2, cells were able to attach well to ECM. However, when cells were grown in media containing UTD2, the cells showed a lower affinity for ECM and were not able to attach; the relative adhesion was 70% at IC50 compared to the controls.
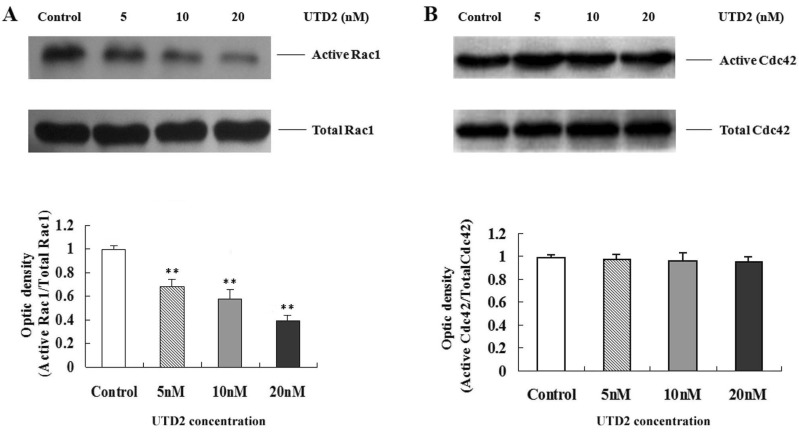
Novel epothilone analog inhibited Rac1 but not Cdc42 activation in breast cancer cells
Comprehensive studies have elaborated the interaction between Rho GTPases and the cytoskeleton [5]. Rac1 and Cdc42 are two well-known critical regulators of the actin cytoskeleton; they play important roles in control of cell motility, migration and adhesion. The activity of both Rac1 and Cdc42 can be tightly regulated through the dynamics of actin and tubulin. To elucidate the mechanism for the UTD2-mediated inhibition of the actin related cellular processes, a pull-down assay was carried out to measure the levels of activated Rac1 and Cdc42 after drug treatment. After 4 hours of drug exposure, the GTP bound active Rac1 and Cdc42 from the total cell lysates were pulled down using the Rac1/Cdc42-binding domain of PAK1; the data was then analyzed by Western blot (Fig. 3). The UTD2 treatment resulted in the inhibition of activated GTP-bound Rac1, while the GTP-bound Cdc42 remained unchanged. Addition of UTD2 yielded no change in the levels of both total Rac1 and Cdc42 proteins. These results reflected differential effects of the novel epothilone analog on Rac1 and Cdc42 activation.
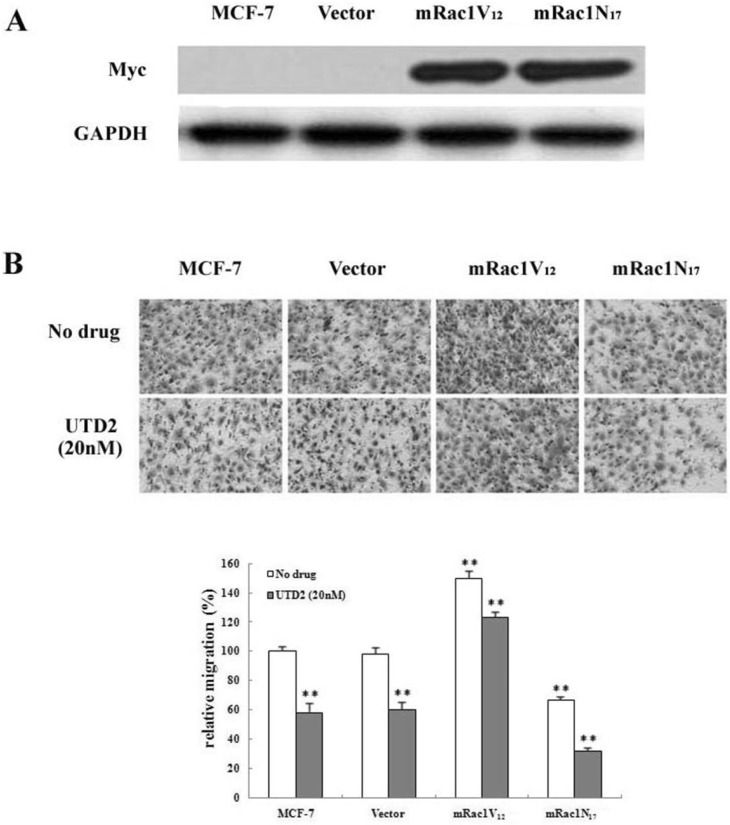
Dominant negative Rac1 potentiated inhibition of breast cancer cell migration by UTD2
Many previous studies using dominant negative mutants of Rac1 have demonstrated that inhibition of Rac1 would lead to inhibition of actin cytoskeleton and cell motility [12]. Therefore, if the effect of UTD2 on cell motility is due to inhibition of Rac1, then it should be expected that the dominant negative mutant of Rac1 will potentiate the UTD2 effect, and constitutively active mutant of Rac1 should antagonize the effect. To confirm these possibilities, MCF-7 cell lines stably expressing myc-tagged Rac1 mutants or empty vector were established. Expression of correct myc-tagged Rac1 mutants in these clones was confirmed by Western blot analysis (Fig. 4A). The effect of UTD2 on migration of these cell lines was tested. As shown in Fig.4B, UTD2-induced inhibition of migration was neutralized in mRac1V12-expressing cells, but was augmented in mRac1N17-expressing cells when compared to parental MCF-7 or vector control cells. Relative migration of mRac1V12-expressing cells was 1.5-fold higher than of MCF-7 cells following UTD2 treatment (Fig. 4B). In contrast, the migration of mRac1N17-expressing cells was about only 2/3 of the control cells (Fig. 4B). These findings demonstrated that UTD2 decreased tumor cell migration via suppressing Rac1 activity.
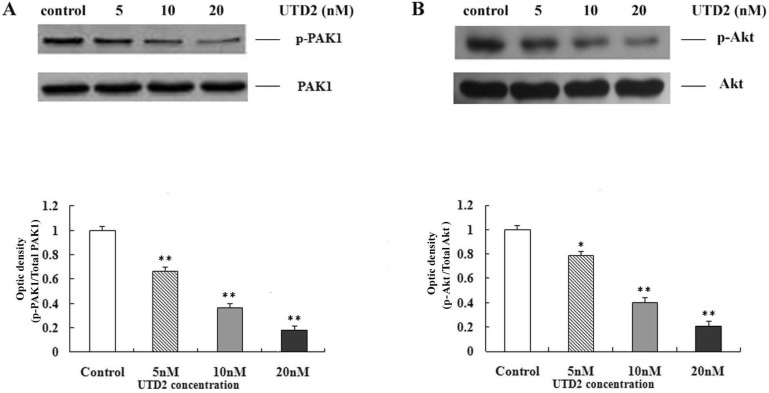
Novel epothilone analog reduced PAK1 phosphorylation
To further elucidate the downstream signaling pathway of Rac1 that attributes to the effects of UTD2 on actin cytoskeleton and cell motility, we tested the effect of UTD2 on the phosphorylation of PAK1 through Western blotting. PAK1 is an effector of Rac1 which has been shown to mediate Rac1 controlled pathways that lead to actin cytoskeletal-related cellular processes, such as cell spreading, cell motility and cell migration. The phosphorylation of PAK1 is essential for its activation and signaling [13]. As shown in Fig. 5A, the novel epothilone analog significantly reduced the phosphorylation of PAK1 in human breast cancer cells (p<0.01). The total expression levels of PAK1 remained the same in MCF-7 cells with or without treatment of UTD2. These results demonstrate that UTD2 efficiently blocked the phosphorylation of PAK1 in human breast cancer cells in a dose-dependent manner.
Novel epothilone analog inhibited activation of cell survival kinase Akt
Akt is a serine/threonine protein kinase that has been implicated in cell migration and is an important regulator of cell survival. It has been previously reported that inhibition of PAK results in attenuation of Akt phosphorylation and decreased tumor cell viability [14]. To determine if the Akt-mediated signaling pathway was affected by UTD2, we tested the effect of UTD2 on the phosphorylation of Akt. As shown in Fig. 5B, 20 nM and 10 nM of UTD2 significantly reduced Akt phosphorylation in human breast cancer cells (p<0.01), and 5 nM of the epothilone only slightly decreased Akt phosphorylation (p<0.05), UTD2 efficiently inhibited activation of cell survival kinase Akt in human breast cancer cells in a dose-dependent manner. The total expression levels of Akt remained the same in MCF-7 cells with or without treatment by UTD2. It has been demonstrated that PI3K/Akt pathway can be downstream of PAK1, and not only is involved in the regulation of cell survival, but also in cell motility [15]. Therefore, our findings indicated that UTD2 may inhibit both cancer cell growth and motility via the Rac1/PAK/PI3K/Akt pathway.
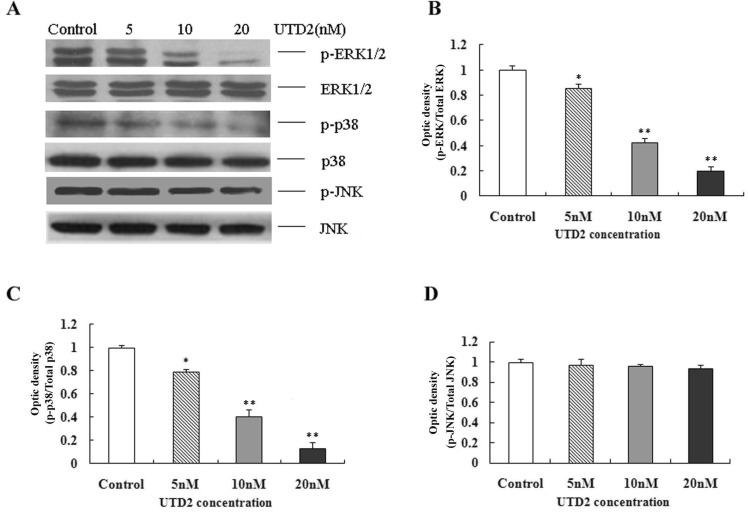
Novel epothilone analog blocked activation of MAPKs
MAPK signaling cascades control mitogenic and apoptotic responses. It has been reported that the three types of MAPKs, ERK1/2, p38 and JNK, could be regulated by Rac1 [14]. To investigate if UTD2 affects MAPK signaling, we measured the phosphorylation levels of ERK1/2, p38 and JNK. Western blot analysis showed that the phosphorylation of ERK1/2 and p38 was reduced to 20% after 24 hours of treatment with 20 nM UTD2 in MCF-7 cells compared to the control cells (Fig. 6B and Fig. 6C). However, JNK phosphorylation remained unaffected (Fig. 6D). These results indicate differential effects of UTD2 on ERK1/2, p38 MAPK and JNK.
Novel epothilone analog affects the synergy between Rac1 and Raf kinase
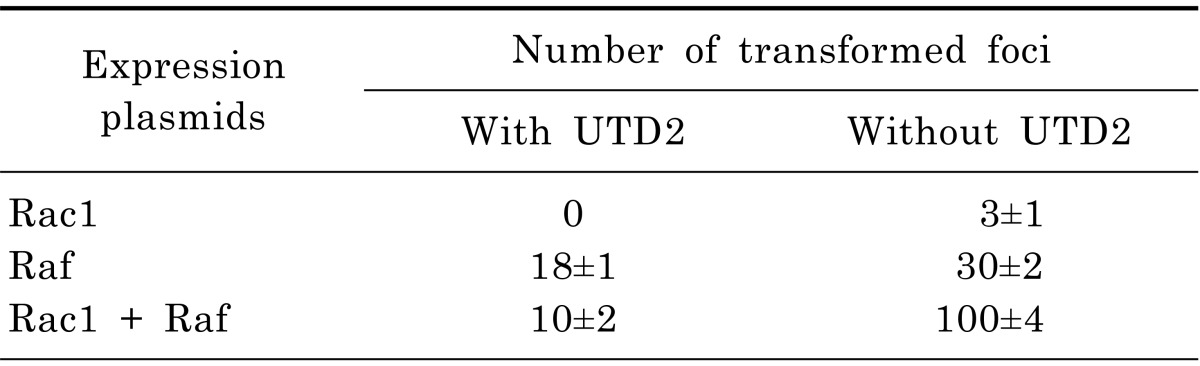
It was previously demonstrated that Rac1 could interact synergistically with Raf kinase to induce transformed foci in a focus formation assay [9]. Raf is an effector of Ras and mediate a signaling pathway that is crucial for both cell proliferation and malignant transformation. To examine the effect of UTD2 on the synergized transformation capacity of Rac1 and Raf, we performed a focus formation assay with NIH3T3 cells, which are sensitive to this assay. Table 2 showed that the number of tansformed foci induced by Rac1 plus Raf were dramatically decreased. Approximately 90% foci were inhibited when cells were exposed to UTD2 compared to untreated cells. UTD2 demonstrated a smaller inhibitory effect on focus formation caused by Raf alone (40% inhibition). UTD2 also abolished focus formation induced by activated Rac1. These findings revealed that UTD2 efficiently affects the synergistic signaling between Rac1 and Raf kinase, and also has a moderate effect on the signaling of Raf. These results further support the notion that UTD2 down-regulates Rac1 activity. These results also suggest that UTD2 can also affect the signaling of Raf kinase, consistent with a inhibitory effect of UTD2 on ERK1/2.
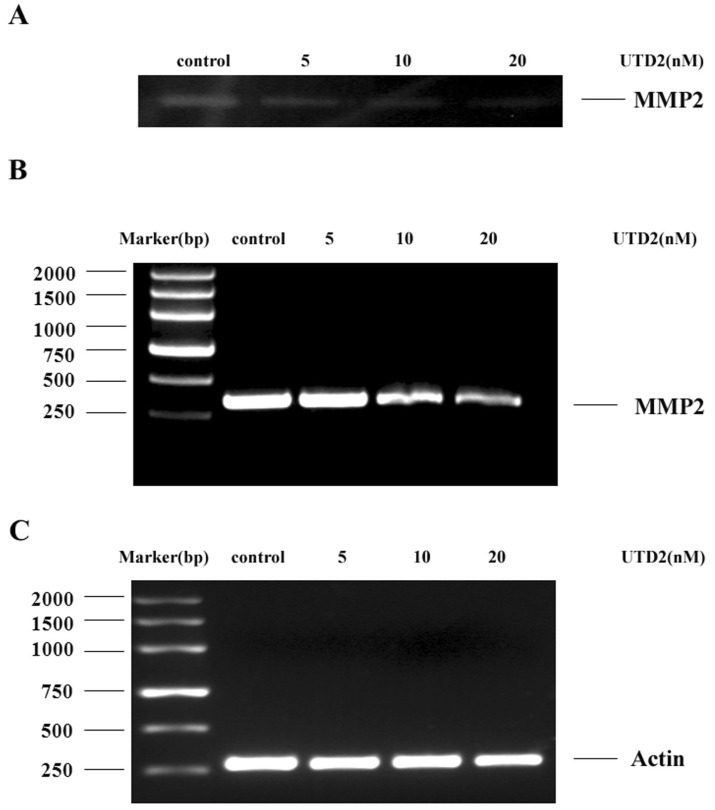
Novel epothilone analog inhibit MMP-2 activity and expression
Matrix metalloproteins (MMPs) play a key role in tumor cell invasion, metastasis, and angiogenesis because they promote the degradation of the extra cellular matrix. Rac1 activity has also been connected to the regulation of MMP-2. To determine whether UTD2's inhibition of Rac1 activation results in the down-regulation of MMP-2, we performed a gelatin zymography assay to analyze the activity of MMP-2 in the UTD2-treated MDA-MB-231 cells. As shown in Fig. 7A, the activity of MMP-2 was clearly reduced after incubation with UTD2 compared to the cells grown in normal culture media. To investigate whether the reduced MMP-2 activity was caused by down-regulation at the gene transcriptional level, we performed a reverse transcription PCR experiment to analyze the mRNA expression of MMP-2 in the MDA-MB-231 cells both with and without the treatment of UTD2. For this experiment, β-actin was used as the control gene. The results indicated that UTD2 significantly down-regulated MMP-2 gene transcription (Fig. 7B). The inhibition of MMP-2 activity and expression was dose dependent.
DISCUSSION
Current clinical data indicated that epothilones, such as Ixempra (ixabepilone), are antineoplastic agents with activities toward different types of breast cancer, including early-stage carcinomas, multi-drug-resistant metastatic breast carcinoma (MBC), and treatment-refractory triple-negative tumors [16]. Understanding the molecular mechanism for the action of epothilones in breast cancer cells may facilitate the clinical development of this novel class of anticancer therapeutics, which may lead to a better understanding of its unique ability to overcome drug resistance and provide new intervention targets/approaches for breast cancer and the treatment of other types of tumors.
Previous studies indicated that the microtubule and actin cytoskeleton cooperate for cancer cell motility [11]. Cell movement is activated via microtubule growth on the leading edge of migrating cells. This microtubule growth is induced when actin-associated microtubules loop and break because of the retrograde flow of polymerizing actin [17]. We have demonstrated that the novel epothilone analog not only interfered with microtubule dynamics, but also disturbed the actin cytoskeletal organization at both sub-toxic drug concentration and 50% growth-inhibiting concentration. This conclusion could be inferred from the cells' lack of movement, the inhibition of wound healing, the observed migration behavior and the interruption of adhesion, which are all actin cytoskeleton-related cellular processes (Fig. 1 and Fig. 2).
Rho GTPases, such as Rac1 and Cdc42, function as molecular switches that regulate the cytoskeleton. Rac1 stimulates the formation of actin extensions in the direction of cell movement and is a crucial regulator for the reorganization of the actin cytoskeleton. The growth phase of microtubule dynamic instability at leading-edge lamellipodia locally activates Rac1 to drive actin polymerization and lamellipodial protrusion which are required for cell migration [18]. Inhibition of microtubule dynamic will, therefore, be likely result in decreased Rac1 activity. We found Rac1 activity to be obviously reduced while microtubule cytoskeleton was disturbed after UTD2 treatment in MCF-7 cells (Fig. 3A), resulting in inhibition of actin cytoskeleton related cellular processes, such as wound healing, migration, adhesion and invasion. In support of a role of Rac1 in molecular mechanism underlying UTD2's effect on actin cytoskeleton, stable expression of dominant negative Rac1 potentiated UTD2's effect on cell migration, and expression of constitutively active Rac1 compromised this effect (Fig. 4). Interestingly, Cdc42 activation was not significantly affected by UTD2 (Fig. 3B), highlighting the differential effects of UTD2 on Rac1 and Cdc42. A possible explanation for this observation is that Cdc42 may not be involved in the control of actin cytoskeleton and cell motility in MCF-7 cells.
One mechanism for Rac1-mediated cytoskeleton reorganization is through its downstream effector PAK1. Emerging evidence suggests that PAK1 has an important role in human breast cancer [19]. Early studies by Kumar and his co-workers indicated that the expression of a kinase-dead PAK1 mutant reduced the invasiveness of MDA-MB-231 breast cancer cells. Conversely, a constitutively active PAK1 mutant promotes MCF-7 cell migration, invasiveness and anchorage-independent growth [20]. This study revealed that PAK1 phosphorylation was significantly reduced after exposure to UTD2, validating the inhibition of Rac1 activation by UTD2.
In the signaling network, MAPK signaling pathways play vital roles in the occurrence of breast cancer. The abnormal activation of MAPKs, including JNK, p38 and ERK, leads to the loss of cell differentiation and apoptosis, resulting in malignant transformation, tumorigenesis, enhancement of invasive ability and ultimately tumor metastasis [21]. p38 has been shown to be involved in the growth factor- and cytokine-induced cell migration; down-regulation of p38 inhibited the motility of breast cancer cells [22]. Previous reports have shown that the activation of p38 can be mediated by Rac1/PAK1 in breast cancer cells [23]. We found that UTD2 not only inhibited PAK phosphorylation, but also affected the activation of ERK1/2 and p38 by reducing the levels of their phosphorylations in the treated MCF-7 cells; however, treatment with this compound did not inhibit the phosphorylation of JNK. Our results indicate differential effects of UTD2 on MAPKs. Since p38 has been reported to be downstream of PAK1 to mediate cell motility, therefore, UTD2 may act on the actin cytoskeleton via the Rac1/PAK1/p38 pathway in breast cancer cells.
It was reported that a critical step for invasion and metastasis of cancer cells is the breakdown of the basement membrane, which requires activation of proteolytic enzymes. Among the group of proteolytic enzymes, MMPs play an important role in tumor invasion and metastasis [24]. MMP-2 (72-kDa gelatinase A) seems to be involved in the initial step of invasion as it hydrolyzes basal membrane type IV collagen and has been frequently associated with the invasive metastatic potential of tumor cells [25]. We clearly showed that UTD2 inhibited the mRNA expression and activity of MMP-2 (Fig. 7) in breast cancer cells. Since the expression level of MMP-2 has been demonstrated to be regulated by the p38 [26], therefore, the Rac1/PAK1/p38/MMP-2 signaling axis might be the critical pathway leading to the inhibition of actin cytoskeleton, migration and invasion of breast cancer cells following UTD2 treatment. In addition, we demonstrated that Rac1N17, a dominant negative mutant and specific inhibitor of Rac1, potentiated UTD2's effect on actin cytoskeleton related cellular processes in MCF-7 cells, further confirming that UTD2 acts on Rac1 to exert its effect on actin cytoskeleton. These data, together with some previous reports, suggest that the Rac1/PAK1/p38/MMP-2 signaling pathway is critical for the UTD2-induced actin cytoskeleton dysfunction.
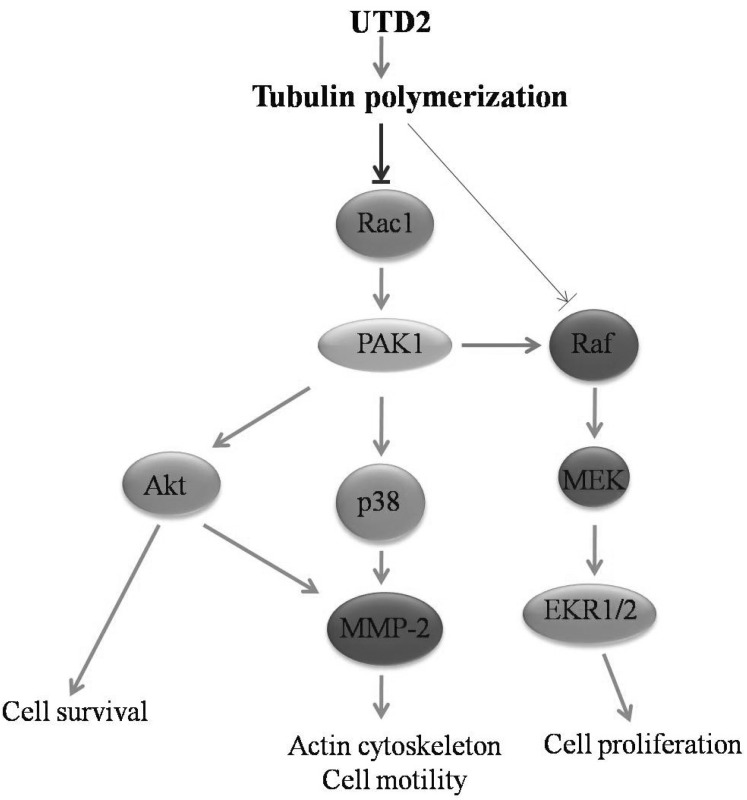
Akt, a serine/threonine kinase, is important for the regulation of cell survival and migration. A previous report has pointed out that Akt potently promoted the invasion of highly metastatic cells by increasing the cells' motility and matrix metalloproteinase production [27]. Our data revealed that UTD2 inhibited the phosphorylation of Akt (Fig. 5B) with a concurrent reduction in the level of expression and activity of MMP-2 in breast cancer cells (Fig. 7). It also has been reported previously that Rac1 may act upstream of Akt [28]. Therefore, our results further suggest that another Rac1-mediated pathway, Rac1/PI3K/Akt/MMP-2 pathway, may be also involved in UTD2-induced actin cytoskeleton dysfunction (Fig. 8).
The Ras/Raf/MEK/ERK pathway is one of the most important pathways in the control of cell proliferations. Previous studies demonstrated that Rac1 plays a critical role in Ras signaling; Rac1 can synergise with Ras effector Raf to enhance oncogenic transformation in focus formation assays, which can be strongly inhibited by dominant negative Rac1. The dominant-negative mutants of Rac1 also inhibit Ras focus formation and revert the morphology of the Ras-transformed cells [29]. In this research we demonstrated that UTD2 also strongly inhibited the synergy between Rac1 and Raf, and moderately reduced the oncogenic potential of activated Raf alone (Table 2) in focus formation assays. Together with the effect on the phosphorylation of ERK1/2, our study indicates that the epthilone analog acts on both the Rac1-mediated pathway(s) and the Ras-Raf-MEK-ERK pathway in MCF-7 cells, which may account for at least part of its antitumor activities.
In this paper, we used IC10 (about 1 nM) and IC50 (about 16 nM) for testing the cytoskeleton related cellular effects and significant changes were detected in cells treated with relative low dose, IC10. We did not use IC10 to examine the effects on signaling pathways, but used 5, 10 and 20 nM instead. We believe that IC10 can actually cause low levels of changes in the signaling molecules which will induce signaling dysfunction, but the Western blotting analysis is not sensitive enough to detect the reduced levels. This explanation can be supported by the effect of epothilones on promoting tubulin polymerization. The mode of action of epothilones is targeting tubulin and promoting tubulin polymerization, which in turn stabilizes microtubule. However, in order to clearly detect an increase in polymerized tubulin induced by epothilones using Western blotting, much higher doses of epothilones than their IC50 (could be 10 folds of that or even higher) need to be used (data not shown).
The research covered in this study indicates that UTD2 exerts multiple effects on cellular processes and signaling pathways regulated by Rac1 GTPase in human breast cancer cells (Fig. 8). It also suggests that the pharmacological inhibition of Rac1 and its downstream signaling molecules may have therapeutic efficacy against tumors. These molecules are also potential biomarkers for epothilone drugs in clinical use. Further analyses of the signaling pathways mediated by these molecules will provide new insight into molecular mechanisms by which the novel epothilone analog UTD2 inhibits tumor growth and metastasis. The actin cytoskeleton, its critical regulators, cell survival factors and matrix metalloproteinases have been implicated in the development of drug resistance in chemotherapy. Therefore, our study may also provide some clues for elucidating how epothilones can overcome drug resistance.

 XML Download
XML Download