

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
ADMA
asymmetric dimethyl arginine
AFGP
alkyl formyl glycosyl pyrrole
AGE
advanced glycation end-products
ALI
arginine-lysine imidazole
AT1R
angiotensin II type 1 receptor
CEL
N-ε-carboxyethyl-lysine
CML
N-ε-carboxymethyl-lysine
3-DG
3-deoxyglucosone
ECM
extra cellular matrix
FFI
2-(2-furoyl)-4(5)-furanyl-1H-imidazole
GLUT1
glucose transporter 1
GOLD
glyoxal-lysine dimmer
ICAM-1
intercellular adhesion molecule-1
IgG
immunoglobulin G
LEC
lens epithelial cell
MGO
methylglyoxal
MOLD
methyl-glyoxal-lysine dimmer
NF-kB
nuclear factor-kB
OST-48
oligosaccharyl transferase-48
PPAR-γ
peroxisome proliferator activated receptor-γ
RAGE
receptor for advanced glycation end-products
ROS
reactive oxygen species
TNF-α
tumour necrosis factor-α
TPS
time to peak shortening
VCAM-1
vascular cell adhesion molecule-1
VEGF
vascular endothelial growth factor
INTRODUCTION
Diabetes and its complications are rapidly becoming the world's most significant cause of morbidity and mortality [1,2]. The adverse effects of persistently elevated plasma glucose levels on the different body parts vary according to the cell types. The cells expressing high levels of the glucose transporter 1 (GLUT 1), such as vascular endothelial cells, are unable to regulate intracellular glucose concentrations and are more susceptible to hyperglycaemia-induced damage. Renal mesangial cells overexpressing GLUT 1 acquire characteristics of the diabetic phenotype, including activation of the polyol pathway and increased extracellular matrix (ECM) synthesis [3]. The complex cascade of events which leads to cellular malfunction in response to high levels of glucose is not fully understood. One of these events is formation of advanced glycation end products (AGEs) [4]. The elevated levels of glucose starts forming covalent adducts with plasma proteins through a non-enzymatic process known as glycation.
Protein glycation reactions leading to AGEs are thought to be the major causes of different diabetic complications [5]. High glucose levels may induce glycation of various structural and functional proteins including plasma proteins and collagen [6]. The non-enzymatic modification of plasma proteins such as albumin, fibrinogen and globulins may be produce various deleterious effects including alteration in drug binding in the plasma, platelet activation, generation of oxygen free radicals, impaired fibrinolysis and impairment in immune system regulation (Fig. 1) [5,7]. On the other hand, the structural impairment in collagen alters the osteoblast differentiation leading to bone remodeling and skeletal fragility [8,9].

Advanced glycation is one of the major pathways involved in the development and progression of different diabetic complications including nephropathy, retinopathy and neuropathy. Tissue and circulating AGE levels are higher in smokers with concurrent increase in inflammatory markers [10]. There is evidence from animal studies that exposure to high levels of exogenous AGEs contributes to renal and vascular complications [11]. AGEs often accumulate intracellularly [12] as a result of their generation from glucose-derived dicarbonyl pre cursors [13]. These intracellular AGEs play important roles as stimuli for activating intracellular signaling pathways as well as modifying the function of intracellular proteins [14]. AGEs accumulate in most sites of diabetes complications, including the kidney, retina, and atherosclerotic plaques [15]. Glycation of proteins interferes with their normal functions by disrupting molecular conformation, altering enzymatic activity, reducing degradation capacity, and interfering with receptor recognition [16]. The mechanism by which glycation altersthe cell functions include denaturation and functional decline of the target protein and lipid, organopathy due to accumulation of AGEs in tissue, activation of receptor-mediated signal pathway in cells, generation of oxidative stress and carbonyl stress [17]. The intermolecular collagen cross-linking caused by AGEs leads to diminished arterial and myocardial compliance, increased vascular stiffness increase, increase in diastolic dysfunction and systolic hypertension [18]. Turk and his co-workers reported that the presence of autoantibodies against serum AGEs are capable of forming AGE-immune complexes in diabetic patients and may play a role in atherogenesis [19]. Glycation-derived free radicals can cause protein fragmentation and oxidation of nucleic acids and lipids [20]. The amino groups of adenine and guanine bases in DNA are also susceptible to glycation and AGE formation [21]. The present review discusses the glycation of proteins such as albumin, fibrinogen, globulins and collagen to form different types of AGEs. Furthermore, the role of AGEs in the pathogenesis of diabetic complications including retinopathy, cataract, neuropathy, nephropathy and cardiomyopathy is also discussed.
Go to : 
MAILLARD REACTION: FORMATION OF ADVANCED GLYCATION END PRODUCTS
The non-enzymatic reaction between the free amino groups of proteins and carbonyl groups of reducing sugars or other carbonyl compounds is known as Maillard reaction [7]. This reaction is subdivided into three main stages: early, intermediate, and late (Fig. 2). In an early stage, glucose (or other reducing sugars such as fructose, pentoses, galactose, mannose, xylulose) react with a free amino group of biological amines to form an unstable compound, the Schiff base which undergoes a rearrangement to a more stable product known as amadori product [22]. In an intermediate stage, the amadori product degrades to a variety of reactive dicarbonyl compounds such as glyoxal, MGO, and deoxyglucosones via dehydration, oxidation and other chemical reactions. In the late stage of glycation, irreversible compoundscalled AGEs are formed through oxidation, dehydration and cyclization reactions. The AGEs are yellow-brown, fluorescent and insoluble adducts that accumulate on long-lived proteins thus impair their physiological functions [23]. AGEs-modified proteins lose their specific functions and undergo accelerated degradation to free AGEs such as 2-(2-furoyl)-4(5)-furanyl-1H-imidazole (FFI), imidazolone, N-ε-carboxy-methyl-lysine (CML), N-ε-carboxy-ethyl-lysine (CEL), glyoxal-lysine dimmer (GOLD), methyl-glyoxal-lysine dimer (MOLD), and others. Moreover, AGEs can also act as cross-linkers between proteins, resulting in the production of proteinase-resistant aggregates [24].
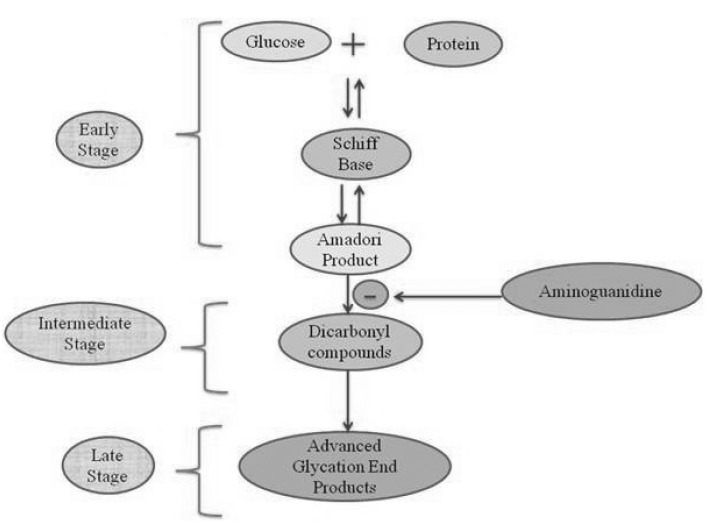 | Fig. 2Formation of advanced glycation end products in three stages i.e., early, intermediate and late stage involving (AGEs). In an early stage, sugars react with a free amino group to form Schiff base which undergoes a rearrangement to a more stable product known as amadori product. In an intermediate stage, amadori product degrades to a variety of reactive dicarbonyl compounds. In the late stage of the glycation process AGEs (irreversible compounds) are formed.
|
In the Maillard reaction, formation of reactive intermediate products during amadori rearrangement is very important. These compounds are known as α-dicarbonyls or oxoaldehydes such as 3-deoxyglucosone (3-DG), methylglyoxal (MGO) (an intermediate product of Maillard reaction) [25]. The 3-DG is formed by non-oxidative rearrangement and hydrolysis of amadori products and from fructose-3-phosphate which is a product of polyol pathway [26]. The formation of AGEs progressively increases with normal aging and has been shown to accumulate in human cartilage, skin collagen and pericardial fluid [27]. Long-lived proteins especially collagens contain numerous lysine, hydroxylysine and arginine residues that are prone to age related accumulation of glycation damage [28]. AGEs are important causative factors for the pathogenesis of diabetes [29], cataracts [30], atherosclerosis [31], diabetic nephropathy [32], and neurodegenerative diseases, including Alzheimer's disease [33]. There are three routes for the AGEs formation: 1) auto-oxidative pathway in which sugars give rise to reactive products by autoxidation, 2) amadori rearrangement, and 3) from the Schiff base. Furthermore, humans are also exposed to exogenous AGEs which are ingested with food. Over a dozen AGEs have been detected in tissues and can be divided into three categories: 1. Fluorescent cross-linking AGEs such as pentosidine and crossline. 2. Non-fluorescent cross-linking AGEs such as imidazolium dilysine cross-links, alkyl formyl glycosyl pyrrole (AFGP) cross-links and arginine-lysine imidazole (ALI) cross-links. 3. Non-cross-linking AGEs such as pyrraline and N-carboxymethyllysine (CML) [29].
Go to :
GLYCATION OF PLASMA PROTEINS
About 50% of plasma proteins are protein albumin, which is the major contributor to osmotic pressure of plasma and assist in the transport of lipids and steroid hormones. Globulins make up 35% of plasma proteins and are used in the transport of ions, hormones and lipids assisting in immune function. Fibrinogen is essential in the clotting of blood and can be converted into insoluble fibrins makes up the 4%. Regulatory proteins such as enzymes, proenzymes and hormones make up less than 1% of plasma proteins. Several types of human and rat serum proteins subjected to non enzymatic glycosylation in vivo. Large proportion of plasma proteins has circulating half lives in human 1~2 weeks [34].
Albumin glycation
Human serum albumin is a single polypeptide chain consisting of 585 amino acid residues having a molecular weight of 66,460 Daltons [35]. It has been used as the model protein for physical biochemists because of its ease of purification. Because of its long half-life time and high concentration as compared to other proteins, it is highly sensitive to glycation [36]. Although there are several lysine and arginine residues in the serum albumin structure, but very few of them can take part in the glycation reaction. It has been known for a long time that human blood proteins like haemoglobin and serum albumin may undergo a slow non-enzymatic glycation, mainly by formation of a Schiff base between ε-amino groups of lysine or arginine residues and glucose molecules in blood [37]. Glycation has the potential to alter the biological structure and function of the serum albumin protein. Once it is glycated, it is less efficient for carrying long chain fatty acid. Clinically albumin may alter the binding of drugs in the plasma at various stages of diabetes [38]. The role of glycated albumin in diabetic retinopathy has also been established [39]. Several in vitro studies have shown the key role of glycated albumin in the platelet activation and aggregation [40]. Glycation of albumin can also affect glucose metabolism in both skeletal muscle and adipocyte cells [41]. In experimental model of adipocyte cell lines, albumin-derived AGE has been shown to trigger the generation of intracellular reactive oxygen species leading to an inhibition of glucose uptake [42]. Furthermore, it is established that glycated albumin, contribute to oxidative modification of intracellular proteins in adipocyte cells [43].
Fibrinogen glycation
Fibrinogen is composed of three pairs of non-identical chains, inter-connected by several disulfide bonds. The protein has a molecular weight of about 34,0000 Daltons which includes a small contribution from the enzymatically attached carbohydrates (4%) and it has a half life of 3~4 days. Investigation has shown that there is no difference in fibrinogen concentration, compaction and kinetics of clot formation between the diabetic subjects and non-diabetic subjects [44]. However, glycation of fibrinogen has been reported to impair fibrinolysis [45] and increase fibrin gel permeability, resulting in formation of a less thrombogenic fibrin network [46]. It has been reported that fibrinogen may be an important target for MGO-derived AGE. MGO-derived modifications of fibrinogen may be a part of the mechanism that leads to enhanced vascular dysfunction and atherosclerosis in diabetics [47].
Immunoglobulin glycation
Glycation of IgG is of special interest due to its influence on the functionality of immunoglobulins and their ability to bind antigens and induce the complement system. Glycation of immunoglobulins has been shown to cause major structural disruptions resulting in their functional disability [48]. IgG constitutes about 75% of the total immunoglobulin in serum. It has four N-terminal amino acids and 80 lysine residues, making it a good target for glycation [49]. An important factor in protein glycation is the half-life of individual proteins; greater the half-life, greater the glycation. IgG with a half-life of 24 days exhibit significant in vivo glycation [50]. The Fc and Fab fragments of the immunoglobulin contain a common domain called the immunoglobulin fold, which is composed of beta sheets and a disulfide linkage. This beta sheet secondary structure is important for immunoglobulin function and any changes to this structure result in loss of antibody activity [51]. Glycated IgG is associated with inflammation and is a target for auto-antibodies in rheumatoid arthritis patients [52]. Among the different AGEs, MGO has been considered as the major contributor of immune suppression in diabetic patients [52].
Go to :
GLYCATION OF COLLAGEN
Collagen is a major component of the ECM and is a prominent target of non-enzymatic glycation [53]. This protein is the longest living protein in higher animals, where it occurs primarily as extracellular, insoluble fibers. These fibers account for the large part of the organic mass of skin, tendon, blood vessels, bone, teeth, cornea and vitreous humor. Collagen also provides the framework for the most of the parenchymal organs, either in its fibrous form or organized in basement membrane. In the body, it is continuously exposed to glucose in vascular and extravascular fluids. Glycation damages the collagen and elastin throughout the body. AGEs changes the collagen properties such as loss of the triple helix solubility and flexibility to increase its rigidity [54]. Non-enzymatic glycation of collagen may exert a negative effect on bone remodeling and interfere with osteoblast differentiation [8,55]. The accumulation of AGEs in bone decreases toughness and increases stiffness, therefore, contributing to skeletal fragility [56]. Some studies have reported that high levels of pentosidine (a fluorescent AGE) have detrimental effects on bone strength [9].
Cross-linked and glycated extracellular proteins (collagen) contribute to aging and diabetes. Type 1 collagen, the major organic component of bone matrix, undergoes a series of post-translational modifications that occur with aging such as non-enzymatic glycation [44]. Some studies concluded that collagen glycation augments the formation and migration of myofibroblasts and participates in the development of fibrosis in diabetes [57]. Studies showed that glycated collagen alters the endothelial cell function and could be an important factor in atherosclerotic plaque development [58]. In vivo, collagen glycation may affect tumor cell metastasis [53]. Recent study reported that arteriosclerosis results from glycation of collagen chains in muscular-type arterioles as glyoxal and MGO form cross-links between collagen fibers [59]. Pageon and his co-workers created a model of reconstructed skin modified by glycation of the collagen used to fabricate the dermal compartment and demonstrated the key role of glycation in skin aging [60].
Go to :
RECEPTORS FOR ADVANCED GLYCATION END PRODUCTS (RAGE)
Interaction of AGEs with their cellular receptors has an important role in the pathogenesis of diabetic complications [61]. RAGE was first described as receptor for AGEs. Many receptors for AGE have been identified, such as lactoferrin, scavenger receptors types I and II, oligosaccharyl transferase-48 (OST-48), 80K-H phosphoprotein, galectin-3, and CD36 [62-64]. RAGE is a multiligand receptor and a member of the immunoglobulin superfamily of cell surface molecules and found on smooth muscle cells, macrophages, endothelial cells and astrocytes. RAGE has been identified as receptor for amyloid-beta peptide (Aβ) and β-sheet fibrils [65] S100/calgranulins [66]; amphoterin [67] and Mac-1 [68]. RAGE is composed of three extracellular domains, which include a V-type that possesses ligand binding properties and two C-type immunoglobulin domains C 1, and C 2, a trans membrane helix and a short cytosolic tail [69]. A fourth trans membrane domain anchors RAGE in the membrane and is connected to a highly charged fifth intracellular domain that mediates interaction with cytosolic transduction molecules.
RAGE acts as a signal transduction receptor for N3-carboxy-methyl-lysine (CML), the major AGE in vivo, and is likely to interact with other AGEs [70]. AGEs bind only to the V domain of RAGE [70,71] and a sustained period of cellular activation mediated by receptor-dependent signaling, leads to inflammation. It is proposed that RAGE activation is largely responsible for the pathogenicity associated with AGEs [72,73]. RAGE can be stimulated not only by AGEs, but other ligands including S100-calgranulins which are a group of pro-inflammatory cytokines, amphoterin, amyloid-β and other fibrillar proteins [74]. Expression of RAGE is enhanced in certain cells during diabetes and inflammation. Interaction of AGEs with RAGE on macrophages causes oxidative stress and activation of nuclear factor-κB (NF-κB) via activation of the p21ras and the mitogen-activated protein (MAP) kinase signaling pathway [75]. NF-κB modulates gene transcription for endothelin-1, tissue factor and thrombomodulin and generation of pro-inflammatory cytokines such as interleukin-1 α (IL-1α), interleukin-6 (IL-6) and tumour necrosis factor-α (TNF-α) [76]. There is also enhanced expression of adhesion molecules including vascular cell adhesion molecule-1 (VCAM-1) and intercellular adhesion molecule-1 (ICAM-1), (Fig. 3) in addition to other effects such as increased vascular permeability. A study showed that NF-κB and heme oxygenase mRNA, the markers of oxidative stress, are activated, following binding of AGEs with RAGE in endothelial cells [77]. During glycoxidative stress, NF-κB activates the production of TNF-α which in turn leads to enhanced ROS production. ROS plays a crucial role in the pathogenesis of type II diabetes, neurodegenerative and cardiovascular diseases [78]. It has been reported that direct exposure of endothelial cells to hyperglycaemic concentrations of glucose increases the formation of ROS, which in turn activates the enzyme NADPH oxidase [79]. The inhibition of AGEs formation is another mode for diabetes treatment, which is not dependent on the control of blood glucose, and would be useful in prevention of certain diabetic complications [80].

Go to :
AGEs AND DIABETIC COMPLICATIONS
AGEs in Diabetic retinopathy
Retinopathy is a serious microvascular complication of diabetes and is the leading cause of blindness in individuals between the ages of 30 and 70 years [81,82] and is characterized by increased proliferation of blood vessels, vascular occlusion, angiogenesis, loss of pericytes from retinal capillaries, microaneurysms, haemorrhages increased retinal capillary permeability, thickening of the capillary basement membrane and infarction affecting the retina of the eye [81]. The potential clinical application of RAGE blockade include the decreased progression of diabetic retinopathy as upregulation of RAGE leads to pro-inflammatory responses by retinal Müller glia cells [83]. AGEs play an important role in the progression of diabetic retinopathy, and leads to dysfunction and death of various retinal cells [84]. The multiple components of the AGE-RAGE axis including signal transduction, formation of ligands, and the end-point effectors can be promising targets for treatments of diabetic retinopathy [85]. Some studies revealed that Müller glial dysfunction during diabetic retinopathy in rats is linked to accumulation of AGEs [86]. Pre-clinical and clinical studies indicate that AGEs including MGO influence various aspects of diabetic retinopathy [87]. Recently, it is evaluated that detoxification of MGOreduces AGEs accumulation which in turn can prevent formation of key retinal neuroglial and vascular lesions [88]. The AGE-RAGE interaction appears to play a central role in the sustained inflammation, neurodegeneration, and retinal microvascular dysfunction occurring during diabetic retinopathy. The reports have revealed that the increased formation of AGEs in the vitreous may be involved in the development of diabetic retinopathy by inducing the production of bFGF (basic fibroblast growth factor) by retinal Müller cells [89]. Studies have provided the strong evidence that the proteins of the human eye are highly susceptible to the formation of AGEs from the reaction of sugars and carbonyl compounds. AGEs progressively accumulate in the lens and retina to adversely affect the vision [90]. Recent findings suggest that N-(ε)-CML, AGEs are the key modulators for the development of nonproliferative retinopathy among type 2 diabetic patients [91]. Some reports suggested that the pathophysiological cascades triggered by AGE have an important role in the onset of the microvascular complications of diabetes including retinopathy [92].
Earlier studies have detected the presence of AGE in the retinal blood vessel walls that contribute towards vascular occlusion and increased permeability of retinal endothelial cells causing vascular leakage [93]. Crosslinking of proteins by AGE in the vessel wall increases vascular stiffness and modification of ECM proteins decreases pericyte adherence [94]. AGE-modified extracellular proteins also cause retinal injury via binding to RAGE [95]. Binding to RAGE activates a variety of signaling pathways leading to increased oxidative stress and synthesis of local growth factors, cytokines and adhesion molecules [96]. AGEs are toxic to AGE receptors possessing pericytes, and damage to pericytes is reported in diabetic retinopathy [97]. Studies have shown that AGEs upregulate RAGE mRNA levels in pericytes and microvascular endothelial cells [98]. This upregulation of RAGE may cause an increase in transduction signals following stimulation by AGEs and this may exacerbate loss of pericytes in diabetic retinopathy. Apoptosis of pericytes usually precedes vascular changes and is a characteristic of early retinopathy [99]. This AGE-induced pericytes death has been associated to signaling through RAGE [100], induction of oxidative stress, inhibition of integrin-mediated protein kinase B/Akt phosphorylation, and reduced survival signaling by PDGF [101].
AGEs also up-regulate ICAM-1 expression in the cultured bovine retinal endothelial cells and contribute to diabetic retinal microvascular leukostasis [102,103]. Exposure of retinal cells to AGEs causes upregulation of the potent mitogen, vascular endothelial cell growth factor (VEGF) by increasing VEGF gene expression. VEGF stimulates angiogenesis and neovascularisation, which are involved in the pathogenesis of proliferative retinopathy. The levels of VEGF in ocular fluid correlate with the activity of neovascularisation in retinopathy and are also associated with the breakdown of the blood-retinal barrier which may be involved in the increased microvascular permeability seen in retinopathy. The reduced retinal perfusion that results from capillary loss leads to ischemia, which is thought to be a stimulus for the breakdown of the blood-retinal barrier and the neovascularisation of the retina [104]. A study has demonstrated an increase in levels of AGEs and IL-6 in the eye vitreous of patients with diabetic retinopathy. AGEs stimulates secretion of IL-6 from the human retinal cells, which induces angiogenesis by increased expression of VEGF [105]. Increased leukocyte adhesion to retinal endothelial cells in experimental diabetes can lead to endothelial cell apoptosis through a Fas- Fas ligand pathway [106]. The increased vascular permeability has been associated to a local increase in VEGF concentration [107]. Within the retina, VEGF is produced by retinal pigment epithelial cells, pericytes, astrocytes, Muller glial and endothelial cells. Local hypoxia and the increased levels of inflammatory cytokines, AGEs and reactive oxygen species that occur in diabetes can induce VEGF gene expression [108].
AGEs in Diabetic cataract
AGEs play a pivotal role in loss of lens transparency, i.e., cataract development [109]. Cataract is a major cause of blindness in developed and developing countries [110]. Progression of cataract is increased in patients with diabetes mellitus [111]. Glycation of eye lens protein has been considered to be one of the mechanisms responsible for diabetic cataract, which is the leading cause of blindness [112]. Recent results have shown that AGEs play an essential role in degenerative changes in lens and approaches towards utilizing low AGEs-content food may be beneficial to delay cataract formation [109]. Some reports have revealed that AGEs accumulate in the lens, and cause vision impairment, cataract. In the lens AGEs induce irreversible changes in structural proteins, which lead to lens protein aggregation and formation of high-molecular-weight aggregates that scatter light and impede vision [90]. It has been shown that AGEs, by altering the surface charge of the protein, lead to conformational change that in turn may affect protein-protein and protein-water interactions and ultimately, lead to decreased transparency of the eye lens [113,114]. The rate of AGEs accumulation is related to the severity of diabetic cataract. Increased glucose levels in the aqueous humor may induce glycation of lens proteins, a process resulting in the generation of superoxide radicals and in the formation of AGEs [30]. Interaction of AGE with RAGE in the epithelium of the lens further increased the O2- and H2O2 generation [115]. In addition to increased levels of free radicals, diabetic lenses show an impaired antioxidant capacity, increasing their susceptibility to oxidative stress [116].
It is demonstrated that protein glycation may be involved in cataract development, by altering protein structure, particularly amino acid composition, and formation of fluorophores through a Maillard reaction [117]. Studies have shown that human lens epithelial cells (LECs) attached to anterior lens capsules express mRNA RAGE and suggested that AGEs may alter cellular functions, which induce mRNAs and proteins associated with fibrosis in LECs. Some studies revealed that AGEs are responsible for the change in the color, opacity of the eye lens and formation of AGEs occurs at a higher rate in cataract lens [118].
AGEs in Diabetic nephropathy
Diabetic nephropathy is defined as a progressive decline in glomerular filtration rate, accompanied by proteinuria and other end-organ complications such as retinopathy [119]. Diabetic nephropathy progresses to end-stage renal disease via a number of stages including normal albuminuria, incipient diabetic nephropathy, micro albuminuria and finally end-stage renal disease [120,121]. Progression to end stage renal disease is enhanced by hyperglycaemia, hypertension and proteinuria, which are all common in diabetes [122,123]. Renal disease in diabetic patients is characterized by haemodynamic (hyperfiltration and hyperperfusion) as well as structural abnormalities (glomerulosclerosis, alterations in tubulointerstitium including interstitial fibrosis) and metabolic changes [124]. Within glomeruli, there is thickening of basement membranes, mesangial expansion and hypertrophy and glomerular epithelial cell (podocyte) loss [125]. Disease progression is also seen in the tubulointerstitial compartment causing expansion of tubular basement membranes, tubular atrophy, interstitial fibrosis and arteriosclerosis.
It has been reported that AGE-RAGE axis play an important role in diabetic nephropathy. Pravastatin has been shown to exert beneficial effects on tubular damage in diabetic nephropathy by inhibiting the AGEs-induced apoptosis and asymmetric dimethylarginine (ADMA) generation tubular cells via suppression of RAGE expression [125]. It is suggested that AGEs play an important role in the pathogenesis of diabetic nephropathy through interacting with RAGE, which activate a series of intracellular signaling pathways. Thus, a novel treatment based on dual-target drugs (AGE inhibitors and RAGE inhibitors) has been proposed which may have potential applications in diabetic nephropathy therapy [126]. Recently it is evaluated that GLP-1 receptor agonist inhibits the asymmetric dimethylarginine (ADMA) (an endogenous inhibitor of nitric oxide synthase) generation in tubular cells and thus, protects against the development and progression of the diabetic nephropathy. ADMA is generated in the tubular cells by AGE-RAGE interaction, thereby suggesting the role of AGEs in diabetic nephropathy [127]. It is demonstrated that AGEs could induce podocyte DNA damage and detachment partly via stimulation of the angiotensin II (Ang II) type 1 receptor (AT1R) [128]. Diabetic nephropathy is characterized by the accumulation of ECM protein in the glomerular mesangium and tubulointerstitium. AGEs may induce an imbalance between the synthesis and degradation of ECM components, leading to the pathologic accumulation of collagens, fibronectins, and laminins [129]. The formation of inter and intramolecular cross-links after the glycation of collagen leads to structural alterations, including changes in packing density [130] and surface charge, manifested by increased stiffness, reduced thermal stability, and resistance to proteolytic digestion [131]. The reduction in collagen pepsin solubility is reflected in a marked increase in acid-insoluble collagen in diabetic tissue [132]. Cell-matrix interactions may also be disrupted by matrix glycation, contributing to changes in cellular adhesion [133], altered cell growth, and loss of the epithelial phenotype. In addition, heterotypic interactions between matrix proteins are disturbed by AGE modifications. The affinity of laminin and fibronectin for type IV collagen and heparan sulfate proteoglycan is decreased after AGE modification [134]. Glycation also inhibits the homotypic interactions required for polymeric self-assembly of type IV collagen and laminin [135]. These changes may be particularly apparent in the glomerular basement membrane, where the induction of chemical cross-links between amines leads to an increase in protein permeability [136].
The expression of extracellular proteins such as fibronectin and types I and IV collagen is increased by AGE in a dose- and time-dependent manner, in the presence [137] or absence of hyperglycemia [138]. This has been considered to be a direct effect via AGE-specific receptors involving activation of the JAK/STAT signal transcription pathway [139], leading to the induction of profibrotic cytokines and growth factors, including TGF-β, PDGF (platelet derived growth factor), and CTGF (connective tissue growth factor; also known as IGF-binding protein-related protein-2) [139]. CTGF is a potent profibrotic agent whose levels are increased in diabetic nephropathy [140]. It is demonstrated that soluble AGE including carboxymethyllysine containing proteins is able to induce the expression of CTGF and fibronectin production in cultured human mesangial cells [141]. Similar changes have also been reported in human dermal fibroblasts, where the AGE-induced upregulation of CTGF is mediated through the RAGE [142]. Excessive ECM production is also compounded by the increased numbers of interstitial fibroblasts, myofibroblasts, and infiltrating macrophages in diabetic nephropathy. RAGEs are expressed in the podocytes of kidneys in both humans and diabetic mice, but not in the mesangial or glomerular endothelial cells and expression of RAGE is upregulated in human patients with diabetic nephropathy [143]. Animal studies have shown that the advanced nephropathy develops in transgenic mice that over-express RAGE in vascular cells. These transgenic mice exhibit increased albuminuria, serum creatinine, mesangial expansion and thickening of the basement membrane [144]. Patients with chronic renal failure have a higher incidence of cancer which may be due to increased intracellular free radical activity capable of damaging DNA following interaction of AGEs with RAGE [145].
AGEs in Diabetic neuropathy
Diabetic neuropathy is a serious complication, affecting both autonomic and peripheral nerves. Diabetic patients with neuropathy present variable symptoms and physical findings, ranging from asymptomatic loss of tendon reflex to severe painful neuropathy. Diabetic neuropathy also causes urinary incontinence, diarrhea, and constipation, impairing the quality of life of diabetic patients. Habib and Brannagan reported that strict glycemic control is one of the therapeutic approach for controlling the diabetic neuropathy [146]. Studies have revealed that AGE-RAGE axis play an important role in the pathogenesis of diabetic foot-associated with diabetic neuropathy [147]. The formation of AGEs by reactive dicarbonyls has been recognized to play an important role in the pathogenesis of sensory neuron damage [148]. It has shown that glycolaldehyde (a precursor of AGEs) at physiological concentration decreases the viability of rat Schwann cells, thus contributes to the pathogenesis and development of diabetic neuropathy [149]. Increased tissue and cellular glucose levels also stimulate glycolytic and polyol pathways in the peripheral nerve [150]. The localization of AGEs has been examined in the peripheral nerve of human diabetic patients and of experimental diabetic animals [151]. In the human diabetic peripheral nerve, CML is present in vascular endothelial cells, pericytes, and basement membrane as well as on the axons and Schwann cells [152]. Studies have shown the increased expression of RAGE in the peripheral nerve in rat by in situ hybridization [153]. In the rat peripheral nerve, RAGE is expressed in endothelial and Schwann cells of perineural and endoneural vessels. The modification of proteins with AGEs causes structural and functional alterations in the peripheral nerve. It is shown that in vitro incubation of neuronal cells and Schwann cells with AGEs induces cell death [154]. The fiber loss in human diabetic peripheral nerve may in part be accounted for by the accumulation of AGE. The modification of neurofilament and tubulin with AGEs may possibly interfere with the axonal transport [155]. The interruption of axonal transport contributes to the development of atrophy and degeneration of nerve fibers. Modification of P0 protein with AGEs may serve as a basis for demyelination of nerve fibers [156].
In vitro experiment demonstrated that oxidative stress enhances glycation of Na+ K+-ATPase (when incubated with glucose) to reduce the activity [157]. The results indicate that glycation of Na+ K+-ATPase may play a role in the reduction in motor nerve conduction velocity as often detected in diabetic human patients and animal models. Microvessels in the peripheral nerve are affected by AGEs. Glycation of collagen and laminin alters the electric charge of the basement membrane to increase the permeability of blood vessels, and cause thickening of the basement membrane. It is reported that nitric oxide (NO), a mediator for vasodilatation, is quenched by AGEs [158]. In addition, AGEs affect the expression level of NO synthase [159] thus, reduces nerve blood flow and induces hypoxia in the peripheral nerve. Because RAGE is expressed in the endothelial cells of peri- and endoneurial blood vessels, it is assumed that the interaction between AGEs and RAGE on the endothelial cells plays a role in the development of peripheral neuropathy [160]. It is shown that the binding of AGEs with RAGE in the endothelial cell activates transcription factors of NF-κB and activator protein-1 (AP-1) to increase the, expression of vascular cell adhesion molecule-1 and cytokines such as tumor necrosis factor and interleukin-6 [72]. It is concluded that formation and accumulation of AGEs in the peripheral nerve involves the development of diabetic neuropathy, directly by affecting structural and functional proteins and indirectly by activating receptors for AGEs [26]. These pathologic processes may affect every cell component in peripheral nervous tissues. It is anticipated that interactions between AGEs and RAGE facilitate endoneural vascular dysfunction, leading to microangiopathy in the peripheral nerve [150].
AGEs in Diabetic cardiomyopathy
Diabetic cardiomyopathy is characterized by myocellular hypertrophy and myocardial fibrosis, which leads to diastolic dysfunction and has high incidence of heart failure in diabetes. Diastolic dysfunction is present in 50~60% of type 2 diabetic patients and is almost present in diabetic patients with microalbuminuria, later progresses into systolic dysfunction. Diastolic dysfunction is related to HbA1c levels, and the most likely reason for this is the accumulation of AGEs in the myocardium [161]. Study have revealed that aminoguanidine is effective in preventing cardiac hypertrophy and arterial stiffening in experimental animal models of diabetes and emphasize the pathogenic role of AGEs in diabetic cardiomyopathy [162]. Methylgyoxal derived AGEs (MG-RAGE) upregulates cardiac RAGE mRNA to trigger the cardiomyocyte contractile dysfunction. MG-AGE elicits the prolongation of time to peak shortening (TPS) and time to 90% relengthening (TR90) in cardiomyocytes and it is associated with mitochondrial membrane potential (MMP) depolarization and reduced GSK-3-beta inactivation in cardiomyocytes [163]. Peroxisome proliferator-activated receptor-gamma (PPAR-γ) activation has been reported to reduce RAGE. Some studies investigated the effects of the PPAR-γ agonist, rosiglitazone, on myocardial expression of RAGE, extent of cardiac fibrosis, and left ventricular (LV) diastolic function in experimental models of diabetes and found that RAGE play an important role in diabetic myocardial fibrosis [164].
AGEs may contribute to the development of heart failure via two pathways: AGEs can affect the physiological properties of proteins in the ECM by inducing the formation of cross-links. AGEs can also cause intracellular changes in vascular and myocardial tissue via interaction with AGE receptors [165]. The diabetic myocyte is exposed to a number of metabolic disturbances that may contribute to contractile dysfunction. Glucose uptake is limited by depletion of glucose transporters, and glucose oxidation is inhibited by high circulating free fatty acids [166], both of which potentially decrease the availability of ATP and thereby reduce contractile function. Uptake of extracellular glucose is regulated by the transmembrane glucose gradient and the activity of glucose transporters in the cardiomyocyte plasma membrane. The GLUT1 transporter, which is localized on the plasma membrane is thought to be a primary mediator of glucose uptake in the heart [167] and its expression is increased within hours of ischemia or induction of hypertrophy. The most abundant glucose transporter in the heart is the GLUT4 transporter. Insulin mediates the translocation of GLUT4 to the plasma membrane from a pool of intracellular vesicles and represents a critical control point by which the net flux of glucose is regulated. A variety of stimuli including hypoxia, ischemia, and cardiac work overload can induce this translocation, thereby increasing glucose uptake and glycolytic metabolism. Upon entry into the cells, free glucose is rapidly phosphorylated by hexokinase to form glucose-6-phosphate (G-6-P), trapping glucose inside the cell. Insulin activates hexokinase in isolated rat hearts and causes the release of hexokinase from the outer mitochondrial membrane, thus increasing the uptake and phosphorylation of glucose [168]. G-6-P can either be used for glycogen synthesis or be channeled down the glycolysis pathway to form pyruvate. It is therefore likely that AGE formation is not only limited to extracellular milieu and the chances of AGE formation intracellularly are very high, as the glucose is converted to G-6-P and trapped inside the cells and is highly reactive in the AGE formation.
Diabetes results in a significant increase in pre-AGE molecules, MGO, AGEs, and RAGE levels in the heart, especially in cardiomyocytes. Diabetes induced decrease in left ventricular contractility is prevented by knockdown of RAGE. AGE-induced cardiomyocyte dysfunction is linked to mitochondrial membrane depolarization, which in turn is prevented by RNA interference knockdown of RAGE expression [163]. Basic fibroblast growth factor is one of the proteins known to be glycated intracellularly. It is also apparent that intracellular sugars and their derivatives may participate in glycation and AGEs formation [12]. During hyperglycemia, a significant rise in the concentration of intracellular sugars occurs, such as glucose-6-phosphate, fructose, and fructose-3-phosphate, all of which are more reactive than glucose. Furthermore, there is increased intracellular accumulation of dicarbonyls such as MGO and glyoxal, potent cross-linking agents. MGO is formed non-enzymatically during anaerobic glycolysis and from oxidative decomposition of polyunsaturated fatty acids and fructose from the polyol pathway [169].
A number of studies have shown that sarcoplasmic reticulum function is compromised in diabetes leading to decreased state of relaxation [170], primarily due to a decrease in sarco-endoplasmic reticulum Ca2+-ATPase (SERCA2a) mRNA and protein expression [171]. A sustained increase in blood glucose leads to increased non-enzymatic glycation of proteins, including SERCA2a and the ryanodine receptor [172]. Exposing the isolated cardiac myocytes to elevated glucose alone results in impaired contractility and calcium handling [173]. Most commonly, AGEs contribute to diabetic complications through (i) formation of cross-links between key molecules in the basement membrane of the ECM, permanently altering cellular structure and architecture, and (ii) interaction of AGEs with RAGE on cell surfaces, altering the signaling cascades and cellular function. Cross-linking of extracellular proteins is a physiologically important phenomenon, which helps to strengthen tissues, without compromising flexibility. AGEs, however, increase the degree of cross-linking over the basal physiological levels, with matrix proteins such as collagen, laminin, vitronectin, and elastin [174]. This excessive cross-linking disturbs the flexibility characteristic of matrix proteins, making them rigid. The increased rigidity decreases the cardiac contractility and induces the diastolic dysfunction in the heart. AGE cross-links on collagen and elastin increase the surface area of ECM to increase the stiffness of the vasculature [14,175]. Glycation of laminin and type I and type IV collagens, key molecules in the basement membrane, reduces adhesion to endothelial cells for both matrix glycoproteins. Studies suggest that AGE formation reduces the binding of collagen and heparin to the adhesive matrix molecule vitronectin [14]. Glycated LDL reduces the NO production and suppresses uptake/clearance of LDL through its receptor on endothelial cells [176]. Another pathway by which AGEs may contribute to the development of diastolic dysfunction is via activation of RAGE [177]. RAGE can effect induction of fibrosis via the up-regulation of transforming growth factor-β (TGF-β) [178]. Glycation also results in increased synthesis of type III collagen, α3(IV) collagen, type V collagen, type VI collagen, laminin, and fibronectin in the ECM, most likely via up-regulation of TGF-β [179]. Petrova and co-workers created transgenic mice that overexpress human RAGE in the heart and analyzed the Ca2+ transients in cultivated cardiac myocytes from the RAGE-transgenic and found that RAGE overexpression reduces the systolic and diastolic intracellular calcium concentration. Thus, it is concluded that the AGE and RAGE could play an active role in the development of diabetes-induced cardiac dysfunction [180].
Go to :
BENEFICIAL EFFECTS OF PHARMACOLOGICAL INTERVENTION IN DIFFERENT DIABETIC COMPLICATIONS
Experimental studies have shown that the pharmacological interventions capable of interfering with AGEs (directly or indirectly) produce beneficial effects in various diabetic complications. Pravastatin reduces tubular damage in diabetic nephropathy by inhibiting ADMA generation in tubular cells and attenuating AGEs-induced apoptosis [125]. GLP-1 receptor agonist inhibits the generation of ADMA generation in tubular cells to attenuate the development and progression of the diabetic nephropathy [127]. Aminoguanidine is shown to prevent cardiac hypertrophy and arterial stiffening in experimental animal model of diabetic cardiomyopathy [162]. Administration of rosiglitazone is shown to produce beneficial effects in cardiac fibrosis and left ventricular diastolic function in experimental models of diabetic myocardial fibrosis by reducing the expression of RAGE on myocardium [164]. Administration of grape seed proanthocyanidins extracts (GSPE) has been shown to be an effective therapeutic agent against diabetic peripheral neuropathic pain by decreasing AGEs [181]. Hesperidin prevents retinal and plasma abnormalities in streptozotocin-induced diabetic rats by inhibiting accumulation of AGEs [182]. A clinical study has reported that epalrestat suppresses the deterioration of diabetic peripheral neuropathy, especially in the lower extremity, by inhibiting the polyol pathway and suppressing the production of AGEs [183]. Stefanska demonstrated that curcumin produces beneficial effects in hepatic fibrosis in type 2 diabetes mellitus by suppressing AGEs-mediated induction of the RAGE gene expression by increasing PPARγ activity [184]. Pyridoxamine, an inhibitor of protein glycation, is shown to produce beneficial effects in relation to microalbuminuria and proinflammatory cytokines in experimental diabetic nephropathy [185]. The extract of Litsea japonica reduces the development of diabetic nephropathy via inhibition of AGEs accumulation in db/db Mice [186]. Korean red ginseng extract alleviates AGEs-mediated renal injury in STZ-induced diabetes [187]. Telmisartan inhibits AGE-induced podocyte damage and detachment in diabetic nephropathy [128] (Table 1).

Go to :
CONCLUSION
The formation of advanced glycation end products appears to be enhanced in the diabetes as a result of hyperglycemia. Increased glycation and accumulation of glycated plasma proteins have an important role in the pathogenesis various diseases. A group of chemical compounds are generated that appears to activate the intracellular signaling pathways and generation of proinflammatory and prosclerotic cytokines which further leads to the development and progression of diabetic complications. There is an important role of RAGE in the pathogenesis of diabetic complications and molecular mechanism of activation of RAGE needs to be investigated. The possibility of reducing glycation and tissue AGEs or by blocking RAGE is an approachable target of delaying or preventing the onset of diabetic complications. Numerous compounds both natural and pharmacological are under investigation for their possible therapeutic potential.
Go to :
 XML Download
XML Download