

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
ACE
angiotensin II-converting enzyme
ACh
acetylcholine
ARB
angiotensin II receptor blocker
Bay-K-8644
methyl-1, 4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoro-methyl-phenyl)-pyridine-5-carboxylate
CA
catecholamines
DMPP
1.1-dimethyl-4-phenyl piperazinium iodide
McN-A-343
3-(m-chlloro-phenyl-carbamoyl-oxy-2-butynyl-trimethyl ammonium chloride
NO
nitric oxide
RAAS
rennin-agiotensinII-aldosterone-system
SHRs
spontaneously hypertensive rats
INTRODUCTION
Fimasartan (BR-A-657; BR-A-657-K; Kanarb®) is a synthetic, nonpeptide, angiotensin II (Ang II) antagonist with an Ang II type 1 (AT1) receptor selectivity, newly developed by Boryung Pharmaceutical Company as an oral treatment for hypertension [1]. It is the eighth drug marketed as a member of the Ang II receptor blocker (ARB) class worldwide [2]. By blocking the effects of Ang II, ARBs relax vascular smooth muscle and thereby promote vasodilation, increase renal salt and water excretion, reduce plasma volume, and decrease cellular hypertrophy. Because of their favorable side effect profile with low potential for drug interaction compared with conventional antihypertensive agents, ARBs deserve increased attention [3,4]. Fimasartan demonstrated no clinically meaningful adverse effects from 20 to 480 mg [5,6]. Moreover, in clinical studies among hypertension patients, fimasartan reduced sitting diastolic blood pressure significantly after multiple once-daily doses of 60 to 120 mg for up to 12 weeks [5]. On the basis of the demonstrated efficacy and safety of fimasartan, the Korea Food and Drug Administration (KFDA) approved it in September 2010. Recently, in a study with eligible adult Korean patients who had mild-to-moderate hypertension, the reduction of sitting DBP after 12 weeks of treatment with fimasartan 60/120 mg was noninferior to that of losartan 50/100 mg. By post hoc comparison, between-group differences in sitting DBP were significant in favor of fimasartan, suggesting superiority to losartan [7].
Generally, the renin-angiotensin II-aldosterone system (RAAS) is an important mediator in the pathophysiology of hypertension, with excessive activity in the RAAS playing a key role in target end-organ damage, such as myocardial infarction, congestive heart failure, coronary artery disease and end-stage renal disease [8]. Thus, this system is a prime target for drugs used in the treatment of hypertension, with two classes of antihypertensive agents targeting the RAAS. The first class, angiotensin II-converting enzyme (ACE) inhibitors (e.g. enalapril, captopril), block the conversion of Ang I to Ang II via inhibition of angiotensin converting enzyme; however, this pathway is only one of several pathways that are involved in the synthesis of Ang II and thus the synthesis of Ang II is only partially blocked [9]. The second class of agents, the Ang II receptor antagonists (ARBs), which includes olmesartan medoxomil, block the RAAS more completely through antagonism of Ang II binding to the Ang II type 1 (AT1) receptor, thereby inhibiting the vasoconstrictor and aldosterone-secreting effects of Ang II [9].
The AT1 antagonist olmesartan blocked both inhibition and facilitation of secretion by Ang II in cultured bovine chromaffin cells [10], and chronic blockade (olmesartan) of the RAAS in rats may decrease the excess sympathetic responses to stress in cardiovascular diseases as well as prevent the likely development of Type II diabetes mellitus [11]. Moreover, we found that other ARBs, such as losartan and olmesartan also inhibit the CA secretion from the perfused rat adrenal gland, which is mediaited through blockade of AT1 recptors [12,13]. In spontaneously hypertensive rats (SHRs), oral administration of AT1 antagonist (candesartan) can effectively block central actions of Ang II, regulating blood pressure and reaction to stress, and selectively and differentially modulating sympathoadrenal responses [14]. Critchley and his colleagues [15] have found that AT1 receptor antagonist candesartan, and the ACE inhibitor ramipril, increased basal CA release from the anaesthetized dog's adrenal gland along with decreases in blood pressure.
Increased activity of the sympathetic nervous system (SNS) accompanied by increased noradrenaline spillover has been implicated in the pathogenesis of essential hypertension in humans [16-18] and in an animal model of this disorder, the SHR [19,20]. The adrenal medulla in combination with the SNS also plays a role in the epigenesis of hypertension in SHR. Hypertension and left ventricular hypertrophy were absent only in sympathectomized SHR that were subjected to adrenal demedullation [21,22]. Studies have also shown that the development of hypertension is attenuated in young SHR subjected to adrenal demedullation [23,24] and that the development of hypertension is restored by chronic adrenaline supplementation [25].
However, there is no evidence of fimasartan's effects on on secretion of catecholamines (CA) from the adrenal medulla so far. To our knowledge, this is the first study that investigates the effects of fimasartan on the CA release in the perfused model of adrenal medulla. Therefore, the aim of the present study was to determine whether fimasartan can inhibit the CA secretion in the perfused rat adrenal medulla of SHRs, and to establish its mechanism of action.
METHODS
Experimental procedure
SHRs, weighing 200 to 300 grams, were anesthetized with thiopental sodium (50 mg/kg) intraperitoneally. The adrenal gland was isolated by the methods described previously [26]. The abdomen was opened by a midline incision, and the left adrenal gland and surrounding area were exposed with the placement of three-hook retractors. The stomach, intestine and portion of the liver were not removed, but pushed over to the right side and covered with saline-soaked gauge pads and urine in the bladder was removed in order to obtain enough working space for tying blood vessels and cannulations.
A cannula, used for perfusion of the adrenal gland, was inserted into the distal end of the renal vein after all branches of adrenal vein (if any), vena cava and aorta were ligated. Heparin (400 IU/ml) was injected into vena cava to prevent blood coagulation before ligating vessels and cannulations. A small slit was made into the adrenal cortex just opposite the entrance of the adrenal vein. Perfusion of the gland was initiated, making sure that no leakage was present, and the perfusion fluid escaped only from the slit made in the adrenal cortex. Then the adrenal gland, along with the ligated blood vessels and the cannula, was carefully removed from the animal and placed on a platform of a leucite chamber. The chamber was continuously circulated with water heated at 37±1℃.
Perfusion of adrenal gland
The adrenal glands were perfused by means of a peristaltic pump (ISCO® pump, WIZ Co., USA) at a rate of 0.32 ml/min. The perfusion was carried out with Krebs-bicarbonate solution of following composition (mM): NaCl, 118.4; KCl, 4.7; CaCl2, 2.5; MgCl2, 1.18; NaHCO3, 25; KH2PO4, 1.2; glucose, 11.7. The solution was constantly bubbled with 95% O2+5% CO2 and the final pH of the solution was maintained at 7.4~7.5. The solution contained disodium EDTA (10 µg/ml) and ascorbic acid (100 µg/ml) to prevent oxidation of catecholamines.
Drug administration
The perfusions of DMPP (100 µM) and Ang II for 1 or 2 minutes and/or a single injection of ACh (5.32 mM) and KCl (56 mM) in a volume of 0.05 ml were made into the perfusion stream via a three-way stopcock, respectively. McN-A-343 (100 µM), veratridine (100 µM), Ang II (100 nM), Bay-K-8644 (10 µM) and cyclopiazonic acid (10 µM) were also perfused for 4 min, respectively.
In the preliminary experiments, it was found that upon administration of the above drugs, the secretory responses to ACh, KCl, McN-A-343, veratridine, Ang II, Bay-K-8644 and cyclopiazonic acid returned to preinjection level in about 4 min, but the responses to DMPP did so in 8 min.
Collection of perfusate
As a rule, prior to stimulation with the various secretagogues, the perfusate was collected for 4 min to determine the spontaneous secretion of CA (background sample). Immediately after the collection of the background sample, collection of the perfusates was continued in another tube as soon as the perfusion medium containing the stimulatory agent reached the adrenal gland. Stimulated sample's perfusate was collected for 4 to 8 min. The amounts secreted in the background sample have been subtracted from that secreted from the stimulated sample to obtain the net secretion value of CA, which is shown in all of the figures.
To study the effect of fimasartan on the spontaneous and evoked secretion, the adrenal gland was perfused with Krebs solution containing fimasartan for 60 min, and then the perfusate was collected for a certain period (background sample). Then the medium was changed to the one containing the stimulating agent or along with fimasartan, and the perfusates were collected for the same period as that for the background sample. The adrenal gland's perfusate was collected in chilled tubes.
Measurement of catecholamines
The CA content of the perfusate was measured directly by the fluorometric method of Anton and Sayre [27] without the intermediate purification alumina for the reasons described earlier [28] using a fluorospectrophotometer (Kontron Co., Milano, Italy).
A volume of 0.2 ml of the perfusate was used for the reaction. The CA content in the perfusate of the stimulated glands by secretagogues used in the present work was high enough to obtain readings several folds greater than the reading of the control samples (unstimulated). The sample blanks were also lowest for perfusates of the stimulated and non-stimulated samples. The content of CA in the perfusate was expressed in terms of norepinephrine (base) equivalents.
Statistical analysis
The statistical difference between the control and pretreated groups was determined by the Student's t and ANOVA tests. A p-value of less than 0.05 was considered to represent statistically significant changes unless specifically noted in the text. Values given in the text refer to means and the standard errors of the mean (SEM). The statistical analysis of the experimental results was made using a computer program described by Tallarida and Murray [28].
Drugs and their sources
The following drugs were used: fimasartan (a gift from Daiichi Sankyo Co., Ltd, Japan), cyclopiazonic acid, acetylcholine chloride, 1.1-dimethyl-4-phenyl piperazinium iodide (DMPP), norepinephrine bitartrate, angiotensin II methyl-1, 4-dihydro-2,6-dimethyl-3-nitro-4-(2-trifluoro-methyl-phenyl)-pyridine-5-carboxylate (Bay-K-8644), veratridine hydrochloride, (Sigma Chemical Co., USA), and (3-(m-chloro-phenyl-carbamoyl-oxy)-2-butynyltrimethyl ammonium chloride [McN-A-343]) (RBI, USA). Drugs were dissolved in distilled water (stock) and added to the normal Krebs solution as required except Bay-K-8644, which was dissolved in 99.5% ethanol and diluted appropriately with Krebs-bicarbonate solution (final concentration of alcohol was less than 0.1%). Concentrations of all drugs are expressed in terms of their molar base.
RESULTS
Effects of fimasartan on the CA secretion evoked by cholinergic agonists
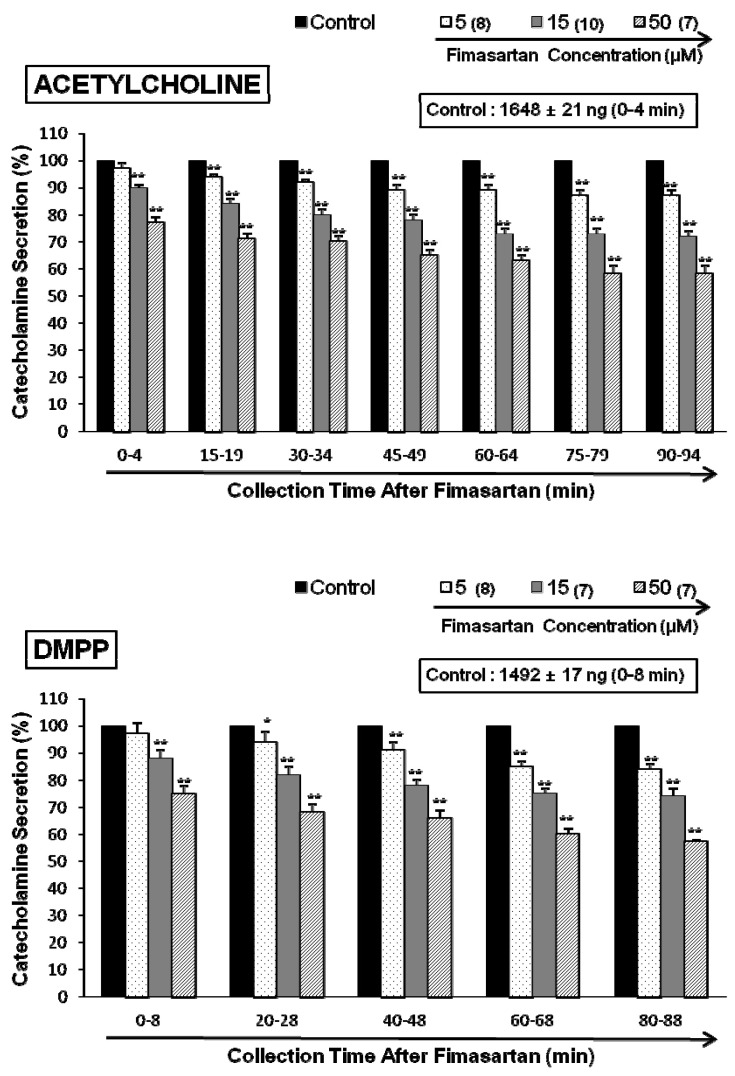
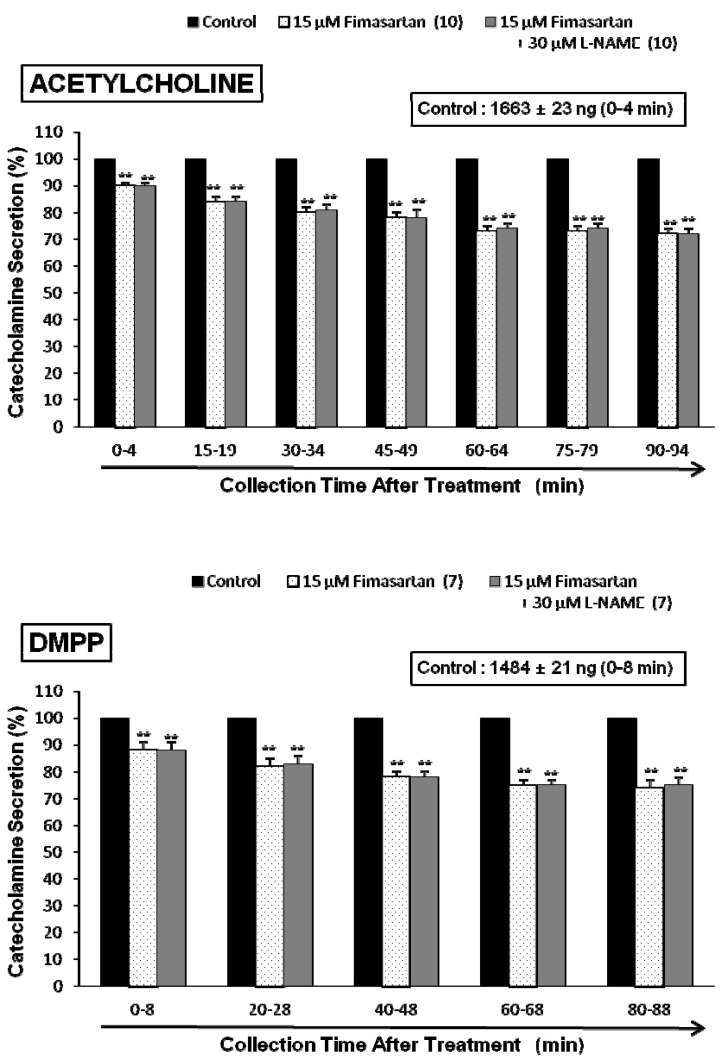
In situ, chromaffin cells are innervated by cholinergic splanchnic nerve fibers. Activation of nicotinic acetylcholine receptors on the chromaffin cells causes membrane depolarization, activation of voltage-gated Ca2+ channels, and influx of Ca2+ that triggers exocytosis of large dense core secretory granules. To mimic this cholinergic stimulation, we used ACh, a neurotransmitter at cholinergic nicotinic receptors, which is released from splanchnic nerve endings. After the perfusion with oxygenated Krebs-bicarbonate solution for 1 hr, basal CA release from the isolated perfused rat adrenal glands amounted to 21±2 ng for 2 min (n=12). Thus, fimasartan was perfused into the adrenal gland for 90 min after the establishment of the control release evoked by each secretagogue. In the presence of fimasartan, ACh (5.32 mM) in a volume of 0.05 ml was given into the perfusion stream at 15-min intervals, and catecholamines were directly quantified using fluorospectrophotometer. As shown in Fig. 1 (upper), Ach evoked robust catecholamine secretion (1,648±21 ng for 0~4 min, mean±SEM), and there was a statistically significant reduction in fimasartan (5~50 µM)-treated adrenal medulla cells in concentration- and time-dependent fashion. ACh-evoked CA secretory responses were inhibited to 58% of the corresponding control release. However, fimasartan itself did not produce any effect on basal CA output from perfused rat adrenal glands (data not shown).
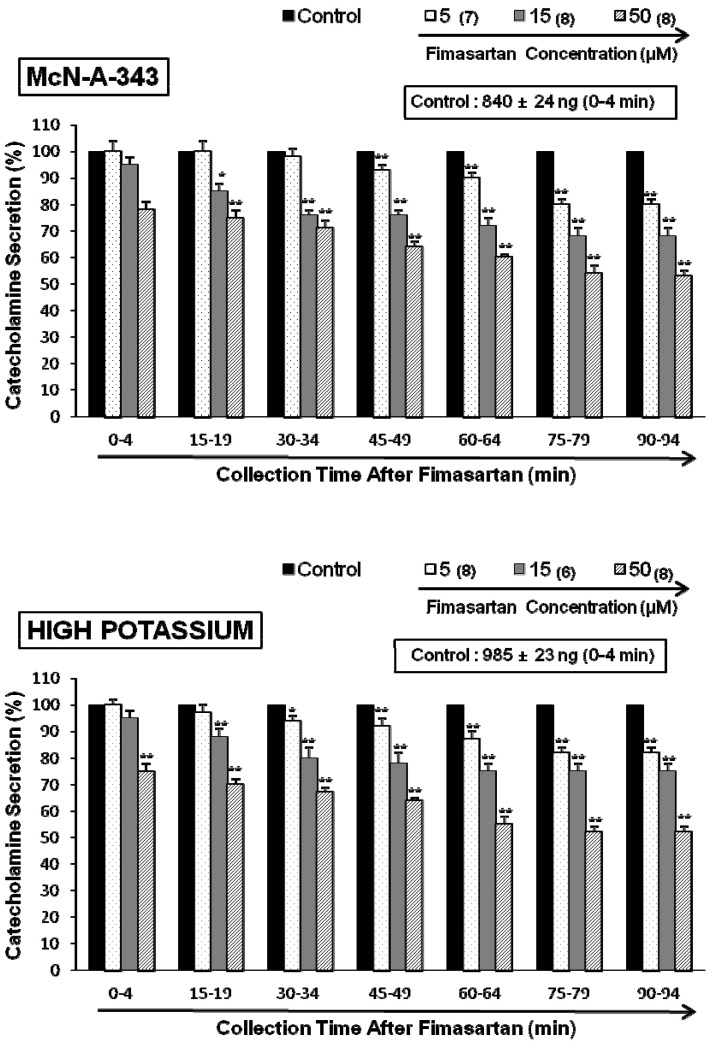
ACh activates both nicotinic and muscarinic receptors on chromaffin cells, but we found similar results when secretion was stimulated with a 1-min application of the selective nicotinic (NN) receptor agonist 1,1-dimethyl-4-phenylpiperazinium iodide (DMPP, 100 µM) in autonomic sympathetic ganglia (Fig. 1-lower). DMPP (100 µM) also evoked a sharp and rapid increase in CA secretion (1492±17 ng for 0~8 min). However, as shown in Fig. 1 (lower), DMPP-evoked CA secretion after treatment with fimasartan was greatly reduced to 57% of the control release (100%). McN-A-343 (100 µM), which is a selective muscarinic M1-receptor agonist [29], perfused into an adrenal gland for 4 min also increased the CA secretion (840±24 ng for 0~4 min). However, in the presence of fimasartan, McN-A-343-evoked CA secretion was markedly reduced to 53% of the corresponding control secretion as shown in Fig. 2 (upper).
Effects of fimasartan on the CA release evoked by KCl, Bay-K-8644, veratridine, cyclopiazonic acid and Ang II
To bypass involvement of cholinergic receptors, we stimulated the cells with 56 mM KCl, which directly depolarizes the membrane potential leading to Ca2+ influx through voltage-gated Ca2+ channels. Previous studies have shown that the secretory response to 30 mM KCl mimics that seen with acute stress [30]. In the present study, fimasartan-treatment significantly depressed KCl-evoked CA secretion compared with control release in a dose-dependent as well as a time-dependent fashion. As shown in Fig. 2 (Lower), high potassium (56 mM) elicited a robust increase in CA secretion (985±23 ng for 0~4 min). However, following the treatment with fimasartan (5~50 µM), high K+ (56 mM)-evoked CA secretion was significantly reduced to 52% of the control secretion at last period (75~94 min).
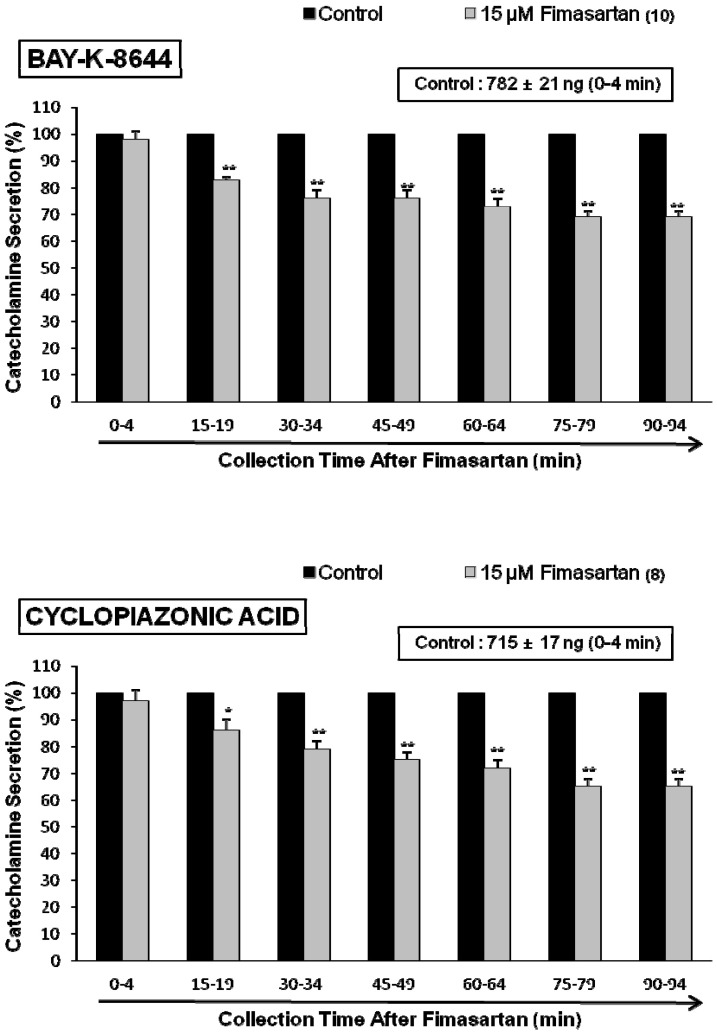
To determine whether cholinergic agonist-evoked Ca2+ influx was altered by fimasartan, we used Bay-K-8644, an activator of voltage-dependent L-type Ca channels. Bay-K-8644 is known to be a calcium channel activator, which enhances basal Ca2+ uptake [31] and CA release [32]. we tried to determine the effect of fimasartan on Bay-K-8644-evoked CA secretion from the isolated perfused rat adrenal glands. Bay-K-8644 (10 µM)-evoked CA secretion in the presence of fimasartan (15 µM) was inhibited maximally to 69% of the control at 75~94 min period as compared to the corresponding control release (782±21 ng for 0~4 min) from 10 adrenal glands as shown in Fig. 3 (Upper).
Cyclopiazonic acid, a mycotoxin from Aspergillus and Penicillium, has been described as a highly selective inhibitor of Ca2+-ATPase in the skeletal muscle sarcoplasmic reticulum [33,34]. The inhibitory effect of fimasartan on cyclopiazonic acid-evoked CA secretion was observed as shown in Fig. 3 (Lower). In the presence of fimasartan (15 µM) from 8 adrenal glands, cyclopiazonic acid (10 µM)-evoked CA secretion was also inhibited to 68% of the control response (715±17 ng for 0~4 min).
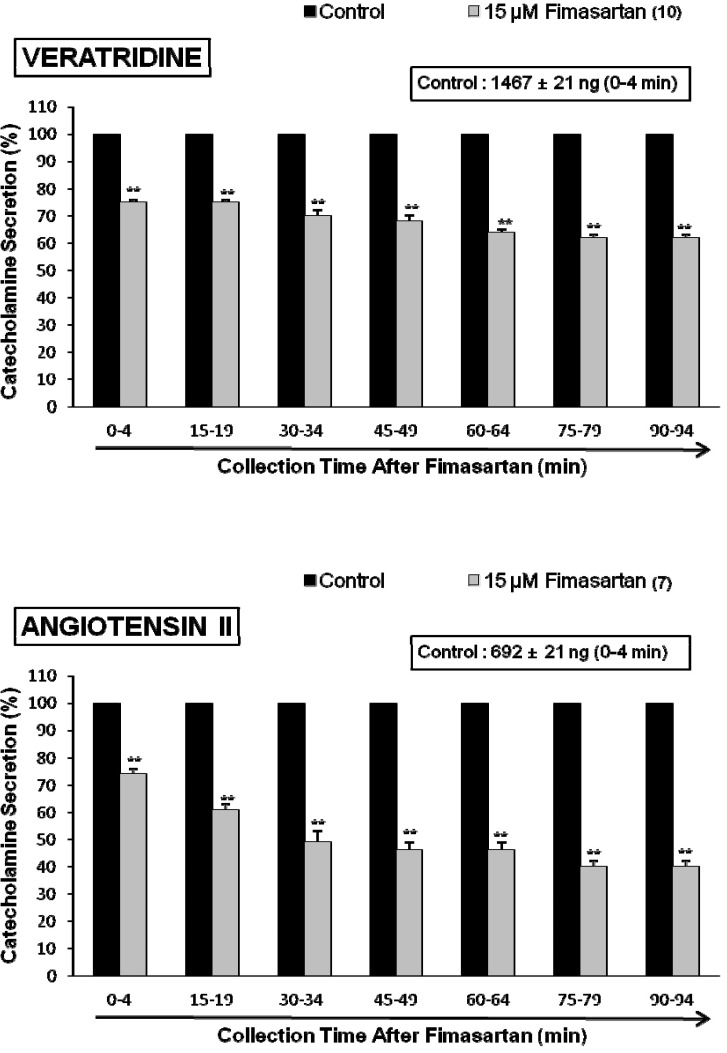
The voltage-dependent Na+ channels consist of the principal α-subunit, which is associated with noncovalently attached β1-subunits, and a disulfide-linked β2-subunit [35]. Previously, it was also found that veratridine-induced Na+ influx mediated through Na+ channels increased Ca2+ influx via activation of voltage-dependent Ca2+ channels and produced the exocytotic secretion of CA in cultured bovine adrenal medullary cells [36]. To characterize the action of fimasartan on voltage-dependent Na+ channels, the effect of fimasartan on veratridine-evoked CA secretion was examined here. As shown in Fig. 4 (Upper), veratridine greatly produced CA secretion (1,467±21 ng for 0~4 min). However, in the presence of fimasartan (15 µM) for 90 min, veratridine (100 µM)-evoked CA secretion was greatly inhibited to 62% of the corresponding control secretion.
Since Hano and his colleagues [37] have suggested that Ang II increase epinephrine release from the adrenal medulla via the AT1 receptors, we examined the effect of fimasartan on Ang II-evoked CA secretion. Ang II (100 nM) significantly increased the CA secretory response (692±21 ng for 0~4 min), while in the presence of fimasartan (15 µM), Ang II-evoked CA secretion was greatly inhibited to 40% of the corresponding control secretion (Fig. 4-lower).
Effect of fimasartan plus L-NAME on CA release evoked by ACh, high K+, DMPP, Ang II, BAY-K-8644 and veratridine
In the present work, fimasartan caused inhibition in the CA secretion by cholinergic receptor stimulation as well as by direct membrane depolarization from the perfused adrenal gland of SHRs. Therefore, in order to establish the relationship between NO and fimasartan-induced inhibitory action on the CA release, we tried to examine the effect of L-NAME on fimasartan-induced inhibitory responses of CA secretion evoked by ACh, DMPP, high K+, Bay-K-8644, verartidine and Ang II.
In the simultaneous presence of fimasartan (15 µM) and L-NAME (30 µM) for 90 min, ACh-evoked CA release was still inhibited to 72% of the corresponding control release (1,663±23 ng for 0~4 min), which is almost the same to the inhibitory effect of fimasartan-treatment alone as shown in Fig. 5 (Upper). Also, the simultaneous perfusion of fimasartan and L-NAME for 90 min did not affect the inhibitory effect of the fimasartan-treatment alone on the DMPP-evoked CA secretion (Fig. 5-Lower). High K+ (56 mM)-evoked CA release in the simultaneous presence of fimasartan (15 µM) and L-NAME (30 µM) for 90 min was also reduced to 75% of the corresponding control release (963±22 ng for 0~4 min), in which there was no difference in comparison to the inhibitory effect of fimasartan-treatment alone on high K+ (56 mM)-evoked CA release (Fig. 6-Upper).
Moreover, in the presence of fimasartan (15 µM) and L-NAME (30 µM), the CA secretory response evoked by Ang II (100 nM) was inhibited to 40% of the corresponding control release (691±24 ng for 0~4 min), which is the same as the inhibitory effect of fimasartan-treatment alone on Ang II-evoked CA secretory response, as shown in Fig. 6 (Lower).
The simultaneous perfusion of fimasartan (15 µM) and L-NAME (30 µM) for 90 min did not affect the inhibitory effect of fimasartan-treatment alone on the CA release evoked by Bay-K-8644 (10 µM, an activator of voltage-dependent Ca2+ channel), as shown in Fig. 7 (Upper). Under coexistence of fimasartan and L-NAME, there was also no difference in the inhibitory effects on veratridine (100 µM, an activator of voltage-dependent Na+ channel)-evoked CA release between fimasartan-treatment alone and combination-treatment (Fig. 7-Lower).
Effect of fimasartan on the level of nitric oxide released from the perfused adrenal medulla
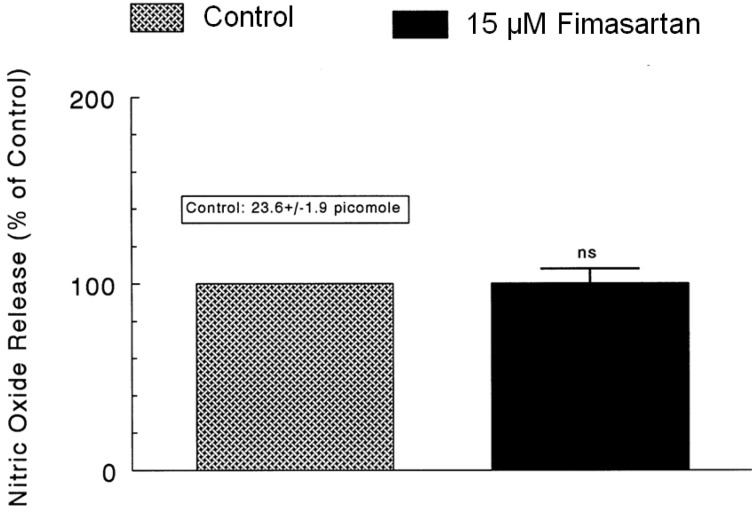
As shown in Fig. 5~7, it has been shown that, in the simultaneous presence of fimasartan and L-NAME (an inhibitor of NO synthase), the CA secretory responses evoked by ACh, DMPP, high K+, Ang II, BAY-K-8644 and veratridine were not affected in comparison with those effects of fimasartan-treatment alone. Therefore, we attempted to determine directly the amount of nitric oxide released from adrenal medulla following the perfusion of fimasartan-containing Krebs-bicarbonate solution. As shown in Fig. 8, the basal level of NO before the loading of fimasartan was 23.6±1.9 picomole. However, 30 min after the presence of fimasartan (15 µM), the level of NO release was not changed in comparison to the control release. Consequently, this result demonstrates that fimasartan fails to affect the release of NO from the rat adrenal medulla.
DISCUSSION
To our knowledge, this is the first study to investigate the effects of fimasartan on the CA release in the perfused adrenal medulla of SHRs. Our rationale for doing so was motivated by two primary goals. First, adrenal medullae (chromaffin cells) are widely used as a model to investigate regulation of Ca2+-channels and neurosecretion [38-40]. Thus, we reasoned this would be a good system in which to evaluate the mechanisms of fimasartan's action. Second, CA, endogenous opioids [41], and other hormones released from adrenal chromaffin cells play central roles in the sympathoadrenal stress response and might contribute to stress-related enhancement of cardiovascular diseases [11]. Thus, we postulated that altered adrenal catecholamineCA release might contribute to hemodynamic stabilization and other beneficial antihypertensive effects of fimasartan. In the present work, we demonstrated that fimasartan inhibits the CA secretion evoked by various secretagogues in the isolated perfused model of SHR's adrenal medulla.
The main findings of the present study have shown that fimasartan can inhibit the CA secretion evoked by cholinergic (both nicotininc and muscarinic receptors) stimulation and direct membrane-depolarization as well as by Ang II from the adrenal medulla of SHRs. This inhibitory effect of fimasartan seems to be mediated by blocking the influx of Na+ and Ca2+ ions through their voltage-dependent ion channels as well as by inhibiting the release of Ca2+ from cytoplasmic store through the blockade of AT1 receptors located on the presynatic membrane of the adrenomedullary chromaffin cells of SHRs without NO production, which is relevant to adrenal cholinergic receptor blockade.
In the adrenal medulla, Ang II releases the CA by a direct action [42], mediated either by circulating Ang II or by the intrinsic RAAS [42-44]. Both AT1 and AT2 Ang II receptors are expressed in the adrenal medulla. In the rat, AT2 receptors predominate, AT1 receptors representing only 5~10% of the total number of Ang II receptors [45]. It appears that AT1 receptor stimulation is most important as a regulatory factor for adrenomedullary CA synthesis and release. First, blocking AT1 is sufficient to inhibit in vivo adrenal CA release by Ang II [46]. Second, pretreatment with an insurmountable AT1 antagonist almost completely abolished the hormonal and sympathoadrenal response to the stress of isolation in unfamiliar metabolic cages [47]. Fimasartan is a selective AT1 receptor antagonist. The concentration that inhibits the binding of [125I]Ang II to the AT1 receptor from rat adrenal cortex by 50% (IC50) was 0.13 nM, compared with 80.0 nM for losartan [1].
In support of the present results, in rats treated with losartan plus chronic stress, plasama epinephrine (EP), norepinephrine (NE), glucose and corticosterone levels was decreased siginificantly compared to chronically stressed rats. These results indicate that chronic blockade of RAAS may decrease the excess sypatheic responses to stress in cardiovascular diseases and prevent the likely development of Type II diabetes mellitus [11]. Previously, we found that losartan inhibits significantly the CA secretion evoked by Ang II in the perfused rat adrenal medulla [12]. Armando and his colleagues [47] also found that pre-treatment with candesartan, an ARB, eliminated the increase in adrenal NE and EP concentrations induced by isolation stress. It has also been shown that acute and chronic stress stimulates the RAAS to increase the levels of Ang II, both in the plasma and brain [48]. Furthermore, olmesartan, by blocking the interaction of Ang II with its AT1 receptor, dose-dependently antagonized Ang II-induced contractions in isolated guinea pig aortic tissue and inhibited the pressor response to intravenously administered Ang II in normotensive rats [49] and also inhibited Ang II-evoked CA secretion in the perfused rat adrenal medulla [13].
Based on previous findings, the present results that fimasartan dose- and time-dependently reduced the CA secretory responses evoked by ACh, DMPP, McN-A-343 and high potassium from the perfused adrenal medulla of SHRs seem to show the blockade by fimasartan of AT1 receptors located presynaptically on adrenomedullary chromaffin cells of SHRs. Moreover, fimasartan, by blocking AT1 receptors, showed superior inhibitory activity in the contraction of isolated rabbit thoracic aorta compared to other ARBs such as losartan and candesartan [1]. In various animal models including renal hypertensive rats, spontaneously hypertensive rats and Beagle dogs, fimasartan effectively reduced blood pressure in a dose-dependent manner following single or repeated oral and intravenous administration [1]. In the present study, as shown in Fig. 4 (lower), fimasartan also greatly inhibited Ang II-evoked CA release from the rat adrenal medulla of SHRs. This finding indicates that fimasartan can inhibit the CA release evoked by various secretagogues as well as by Ang II through the AT1 receptor blockade. Also, it has been shown that CV-11974, an AT1 receptor antagonist, almost completely suppressed Ang II-induced increases in epinephrine released from the isolated rat adrenal gland whereas PD123319, an AT2 receptor antagonist had no significant effects on AT II-induced increases in epinephrine release [37]. Critchley and his colleagues [15] have also found that candesartan, another AT1 receptor antagonist, blocked the CA release induced by Ang II from the anaesthetized dog's adrenal gland along with decreases in blood pressure. These results suggest that Ang II increases epinephrine release from the adrenal medulla via the AT1 receptor.
In the present work as shown in Fig. 5~7, in the simultaneous presence of L-NAME (an inhibitor of NO synthase), the inhibitory effects of fimasartan on the CA secretory responses evoked by ACh, DMPP, high K+, Bay-K-8644 and veratridine as well by Ang II from the perfused adrenal glands of SHRs were not affected in comparison to the group of fimasartan-treated alone. This result suggests that the inhibitory effect of fimasartan is not associated with NO production due to activation of NO synthase in the adrenal medulla. The functional role of AT2 receptors in vascular control has not been well defined. Generally, the adrenal medulla possesses characteristic postganglionic sympathetic neurons, and the presence of neuronal NO synthase (nNOS) has been demonstrated [50-53]. In vitro studies using NOS inhibitors and NO donors were performed to examine the role of NO in modulating CA secretion from the adrenal medulla but the results remain controversial. It has been reported that the NOS inhibitor, L-NAME enhances K+-stimulated CA secretion in cultured bovine chromaffin cells [54] and that sodium nitroprusside inhibits ACh-induced CA secretion in bovine chromaffin cells [55]. These studies suggest that NO may play an inhibitory role in the control of CA secretion. Moreover, the presence of endothelial cells has been reported to inhibit the K+-induced or the nicotinic receptor agonist DMPP-induced CA secretion in cultured bovine chromaffin cells [54], suggesting that not only nNOS but also eNOS may play roles in modulating adrenal CA secretion. In vivo studies suggest that the AT2 receptor causes vasodilatation by stimulating NO, bradykinin, or eicosanoid production in renal arterioles [56,57]. Moreover, mice lacking the AT2 receptor have slightly elevated systolic blood pressures at baseline, and higher pressure sensitivity to Ang II compared with that of wild-type controls [58]. This hints at a role of AT2 receptors for vasodilatation. In contrast, Ang II has been shown to release NO via the AT1 receptor in endothelial cells and in rings from the carotid artery [59,60]. In addition, AT1 receptor inhibition blunts Ang II-induced NO release in larger, isolated perfused renal resistant arteries [61]. This suggests an AT1 receptor-mediated activation of NO systems in renal resistance vessels. Based on previous reports, the results of the present study demonstrate that NO-production is not contributed to the inhibitory effect of fimasartan.
The nicotinic receptor is a neurotransmitter-gated cation-conducting ion channel that is opened by binding of agonists such as ACh and DMPP [62]. The opening of this channel triggers Ca2+ uptake and the CA secretion from chromaffin cells [36]. To determine if the inhibition of both ACh- and DMPP-stimulated secretion by fimasartan was due to an effect on the activity of the nicotinic receptor, the effect of fimasartan, an AT1-selective antagonist, on both ACh- and DMPP-stimulated CA secretion was examined. As shown in Fig. 1, treatment with fimasartan greatly inhibited ACh- and DMPP-evoked CA secretion, reducing to 58% and 57% of the control release, respectively. The present data are similar to the result that chronic immobilization stress increased plasma glucose, NE, E and corticosterone levels in rats, and that losartan significantly prevented these increments induced by chronic stress when given before the stress regimen [11]. Therefore, it is likely plausible that fimasartan can alter the activity of both nicotinic receptors and voltage-sensitive Na+ channels.
In the present study, fimasartan inhibited the CA secretory responses by high potassium, a direct membrane depolarizer, as well as by Bay-K-8644, an activator of voltage-dependent L-type Ca2+ channels, which facilitates the influx of Ca2+ into the cells. The observation that fimasartan inhibited the CA secretion evoked by Bay-K-8644 was surprising, as Takekoshi et al. [63] have reported that removal of external Ca2+ significantly suppressed either AngII plus CV-11974 (AT1 antagonist, 100 nM; which simulates specific AT2 stimulation) or CGP 42112 (AT2 agonist)-induced CA secretion in cultured porcine adrenomedullary chromaffin cells. It is unclear how the blockade of AT1 receptors results in the inhibition of the CA secretion seen in these cells. In the present work, the simplest interpretation is that the decrease in Ca2+ uptake by fimasartan is responsible for the observed inhibition of the CA secretion. However, such an interpretation is complicated by the complexity of the relationship between the CA secretion and intraceIlular free Ca2+ levels. Both the intracellular location of the Ca2+ level increase [64,65] and the magnitude of the Ca2+ level increase [66] can affect the relationship between intracellular free Ca2+ levels and secretion. Holz et al. [66] have reported that when Ca2+ uptake is large, changes in Ca2+ uptake resulted in less than proportional changes in CA secretion. Consequently, although the decrease in Ca2+ uptake (influx) into the adrenal chromaffin cells may explain the decrease by fimasartan in CA secretion, it is still unclear whether this is the only or even most important factor contributing to the inhibition of CA secretion by fimasartan. However, in view of the results obtained from the present study, it is felt that the voltage-sensitive Ca2+ channels located on adrenomedullary chromaffin cell membrane of SHRs could be the target site for fimasartan-mediated inhibition of CA secretion.
In the present study, fimasartan also inhibited the CA secretory responses evoked by cyclopiazonic acid, which is known to be a highly selective inhibitor of Ca2+-ATPase in skeletal muscle sarcoplasmic reticulum [33,34]. Therefore, it is felt that the inhibitory effect of fimasartan on the CA secretion evoked by various cholinergic secretagogues as well as by Ang II may be associated with the mobilization of intracellular Ca2+ in the chromaffin cells. This indicates that fimasartan-induced blockade of AT1 receptors causes an inhibitory effect on the release of Ca2+ from the intracellular pools induced by stimulation of muscarinic ACh receptors, which is weakly responsible for the secretion of CA. In the present work, fimasartan time- and concentration-dependently inhibited the CA secretion evoked by McNA-343, a selective muscarinic M1-agonist. This fact suggests a new concept that fimasartan can modulate the CA secretory process induced by activation of muscarinic M1-receptors as well as neuronal nicotinic receptors in the adrenal medulla of SHRs. In supporting this finding, it has been shown that cyclopiazonic acid easily penetrates into the cytoplasm through the plasma membrane and reduces Ca2+-ATPase activity in sarcoplasmic/endoplasmic reticulum, resulting in increase in the subsequent Ca2+ release from those storage sites and thereby increase of Ca2+-dependent K+-current [67]. Moreover, in bovine adrenal chromaffin cells, stimulation of muscarinic ACh receptors is also proposed to cause activation of phosphoinositide metabolism, resulting in the formation of inositol 1,4,5-trisphosphate, which induces the mobilization of Ca2+ from the intracellular pools [64,68]. However, in the present study, it is uncertain whether the inhibitory effect of the fimasartan on Ca2+ movement from intracellular pools is due to their direct effect on the PI response or the indirect effect as a result of AT1 receptor blockade by fimasartan. Based on these previous results, this finding of the present work suggests that fimasartan can regulate the CA secretion evoked by muscarinic M1-receptor stimulation in the rat adrenal medullary chromaffin cells. Furthermore, Ang II is a secretogogue for CA release that is believed to be mediated through IP3 production by AT1 [46,69]. Indeed, Wong and his colleagues [46] demonstrated that AngII-induced CA release is mediated by AT1 in the rat adrenal medulla. AT1-mediated phospholipase C activation and subsequent IP3 formation may increase cytosolic Ca2+ levels by releasing Ca2+ from intracellular storage, with subsequent activation of CA release [45]. Indeed, it has been shown that addition of IP3 to permeabilized bovine chromaffin cells releases intracellular Ca2+ [70]. Furthermore, addition of Ca2+ to permeabilized bovine chromaffin cells was reported to cause CA secretion [71].
Collectively, the main findings of the present study have demonstrated that fimasartan can inhibit the CA secretion evoked by cholinergic (both nicotininc and muscarinic receptors) stimulation as well as by Ang II from the adrenal medulla of SHRs. This inhibitory effect of fimasartan seems to be mediated by blocking the influx of Na+ and Ca2+ ions through their voltage-dependent ion channels as well as by inhibiting the release of Ca2+ from cytoplasmic store through the blockade of AT1 receptors located on the presynatic membrane of the adrenomedullary chromaffin cells of SHRs without NO production, which is relevant to adrenal cholinergic receptor blockade.


 XML Download
XML Download