

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
K2P
two-pore domain K+ channel
TWIK
Tandem P domain weak inwardly rectifying K+ channel
TASK
TWIK-related acid-sensing K+ channel
TREK
TWIK-related channel
CDK
cyclin-dependent kinase
qRT-PCR
quantitative real-time reverse transcription-polymerase chain reaction
siRNA
small interfering RNA
Kv
voltage-dependent K
KCa
Ca2+-activated K+
INTRODUCTION
K+ channels regulate membrane potential and may affect the oncogenic state of cells through cell cycle regulation, migration, invasiveness and Ca2+ signaling [1]. Nevertheless, the involvement of changes in membrane potential in cell growth mechanisms remains controversial [1]. Several K+ channels are expressed in a variety of malignant cancer cells [1-4]. The type of K channels related to proliferation include voltage-gated channels, such as KV1.3, KV10.1 (Eag1), KV1.5, KV3.1, KV3.4, and KV11.1 (HERG), Ca2+-dependent K+ channels, such as KCa1.1 and KCa3.1 (also called IKCa), ATP-dependent K+ channels and inward rectifiers [1,4].
In addition, some two-pore domain K+ channels (K2P), which are divided into TWIK, TREK, TASK, TALK, TRESK and THIK, are also expressed in various tissues [5]. K2P channels have been known to regulate resting membrane potential and are modulated by antidepressants, antipsychotics, volatile anesthetics, flavonoids, hypoxia, temperature, hydrogen, polyunsaturated fatty acids, cell volume (membrane stretch), and lysophospholipids [5,6]. Therefore, similar to other K+ channels, K2P channels might play an essential role in cancer cells [1,4,5]. Supporting data show that TASK3 was revealed as an oncogene that is frequently overexpressed in breast, lung, colon, and metastatic prostate cancers [7]. TREK1 was also reported as a novel tumor marker in prostate cancer cells [8]. More recently, the presence of the TREK2 channel was reported in normal human ovaries and ovarian cancer [9]. Nonetheless, sufficient data on TREK2 in cancer cells is limited.
More than 90% of bladder cancers are transitional cell carcinomas (TCC), and one third of superficial tumors progress to muscle invasive cancer [10,11]. In addition, the recurrence rate of bladder cancer is relatively high compared to other cancers, and diagnosis is difficult due to low sensitivity biomarkers [10,11]. Furthermore, the mechanism of bladder cancer development and progression, especially in terms of ion channels, is not well established. There are only a few reports showing that the growth modulation mechanism involves ion channels in bladder cancer cells, including BKCa in 253J cells [12], induction of apoptosis by TRPV2 in T24 cells [13], and cell cycle arrest by capsaicin in RT4 cells [14].
In the present study, we investigated the involvement of the TREK2 channel in bladder cell growth. The primary objective of the study is to determine whether TREK2 is functionally expressed in bladder cancer cells, and if TREK2 is involved in cell growth. To achieve this aim, we used electrophysiological and molecular biological methods.
METHODS
Cell lines and culture conditions
The human bladder cancer cell line 253J cells were maintained in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 100 units per ml penicillin, and 100 µg per ml streptomycin in a humidified incubator at 37℃ with 5% CO2. EJ, HT1376 and TCCSUP cells were maintained in DMEM, and T24, 5637 and J82 cells were maintained in RPMI-1640 supplemented with 10% FBS.
Electrophysiological recording and chemicals
Electrophysiological recording was performed in whole cell configurations and single channel recordings [15] using a patch clamp amplifier Axopatch 200B (Axon Instruments, Foster City, USA). Whole cell patch pipettes (Harvard Apparatus, Edenbridge, UK) were prepared at a resistance of 2~3 megaohms. To measure resting membrane potential, the current was clamped (I=0) in a whole cell patch configuration. The solution for the High-K+ pipette (internal solution) was composed of 150 mM KCl, 1 mM MgCl2, 5 mM Mg-ATP, and 2 mM EGTA (pH 7.2), and the normal Tyrode's (NT) bath solution (external solution) was prepared as 143 mM NaCl, 5.4 mM KCl, 0.5 mM NaH2PO4, 0.5 mM MgCl2, 1.8 mM CaCl2, 5 mM HEPES, and 10 mM glucose (pH 7.4). For single TREK2 channel recordings, the pipette and bath solutions were applied symmetrically with 150 mM KCl, 1 mM MgCl2, 10 mM HEPES, and 5 mM EGTA (pH 7.2). The recorded signal was filtered at 1 kHz using an 8-pole Bessel filter (-3 dB: Frequency Devices) and transferred to a computer using the Digidata 1322A interface (Axon Instruments, Union City, CA) at a sampling rate of 2 kHz. Single-channel currents were analyzed with the pClamp program version 9.02 (Axon Instruments, Union City, USA). Data were described as channel activity (NPo). N is the number of channels in the patch, and PO is the probability of a channel being open. The current tracings shown in the figures were filtered at 1 kHz. All recordings were performed at room temperature (22~24℃).
Isolation of total RNA, reverse transcription-polymerase chain reaction (RT-PCR) and quantitative real-time RT-PCR (qRT-PCR)
Total RNA isolation was carried out with a MN NucleoSpin RNAII Kit (Macherey-Nagel, Germany), followed by first-strand cDNA synthesis with an iScript cDNA synthesis kit (Bio-Rad, Hercules, USA), according to the manufacturers' protocols. RT-PCR was performed using 2 µl of cDNA in a 30 µl reaction containing 0.4 mM of each primer. The cycling conditions were as follows: 40 cycles at 96℃ for 15 s, 57℃ for 30 s, and 72℃ for 1 min. An aliquot (20 µl) of the RT-PCR product was analyzed on a 2% agarose gel prepared in Tris-acetic acid-EDTA (TAE). The identity of each of the PCR products was confirmed by sequence analysis. The sequences for primers were as follows: TREK1 (Accession No; AF129399), sense 5' GGATTTGGAAACATCTCACCACGCACA 3', antisense 5' GATCCACCTGCAACGTAGTC 3' (355 base pair [bp]); TREK2 (Accession No; AF279890), sense 5' AAGCATGGGCAGGGTGCGTC 3', antisense 5' TCCGGCTCCCGGTCTTTGGT 3' (291 bp). Quantitative real-time RT-PCR (qRT-PCR) was performed using CFX 96 real-time thermal cycler (Bio-Rad, Hercules, USA), and the relative amount of TREK2 "target" in bladder cancer cells was normalized to the endogenous control "GAPDH". The cycling conditions were as follows: 40 cycles at 95℃ for 10 s, 60℃ for 10 s and 72℃ for 30 s with SYBR green (Bio-Rad, Hercules, USA). Oligonucleotide primers for qRT-PCR to detect human TREK2 were forward 5' CACCTCCAGACTCACCAA 3' and reverse 5' TCCTCCTCTTTCTTCTCCTC', and forward 5'ACCAGGTGGTCTCCTCTGAC 3' and reverse 5' TGCTGTAGCCAAATTCGTTG 3' were used to detect GAPDH. The relative gene expression was analyzed using CFX manger software system version 3.0 (Bio-Rad, Hercules, USA).
Downregulation of TREK2 expression by small interfering RNA
TREK2 small interfering RNA (siRNA), negative control siRNA and fluorescein isothiocyanate (FITC)-labeled siRNA were purchased from Bioneer (Daejon, Korea). The TREK2 siRNA sequences were as follows: sense 5' CAUCUUUGGGAAAGCAUU 3' and antisense 5' AAUGCUUUUCCCAAAGAU G 3'. The negative control siRNA sequences were as follows: sense 5' CCUACGCCACCAAUUUCG 3' and antisense 5' ACGAAAUUGGUGGCGUAGG 3'. Lipofectamine RNAiMax (Invitrogen, Carlsbad, USA) was used for siRNA transfection. Cells were incubated for 72 hours after transfection.
Proliferation assay
Cell proliferation was monitored using an XTT cell proliferation assay kit (Biological Industries Israel Beit-Haemek Ltd., Kibbutz Beit-Haemek, Israel) following the protocols described by the manufacturer. Briefly, 253J cells were prepared at a density of 10,000 cells/ml in a 96-well plate containing 2% FBS, 100 units per ml penicillin, and 100 µg per ml streptomycin. Sample absorbance was measured with a SpectraMax M5e spectrophotometer (Molecular Devices, Sunnyvale, USA) at a wavelength of 450~500 nanometers. Reference absorbance to measure non-specific readings was measured at a wavelength of 630~690 nanometers.
Western blot assay
Bladder cancer 253J cells and TREK2 transiently transfected CHO cells were homogenized in lysis buffer (50 mM HEPES, pH 7.5, 150 mM NaCl, 100 mM NaF, 10% glycerol, 1% Triton X-100, 200 µM Na3VO4, 1 mM phenylmethylsulfonyl fluoride, 1 µg/ml aprotinin, and 1 µg/ml leupeptin). Samples containing 40 µg total protein were separated by 10% sodium dodecyl sulfate polyacrylamide (SDS-PAGE) gel, transferred onto Immobilon-P membranes (Millipore Corporation, Bedford, USA) and incubated with rabbit polyclonal anti-hKCNK10 (TREK2) (concentration of 1:1,000, Alomone labs, Jerusalem, Israel) antibodies. Proteins were visualized using polyclonal goat anti-rabbit IgG conjugated to horseradish peroxidase (concentration of 1:5,000, Santa Cruz Biotechnology, Santa Cruz, USA) and enhanced chemiluminescence (ECL) reagent (Amersham Biosciences, Piscataway, USA) on a Lumi-Imager F1 scanner (Roche Applied Science, Indianapolis, USA). Images were analyzed using Lumi-analyst software version 3.1 (Roche Applied Science, Indianapolis, USA). Antibodies used to confirm the expression of cell cycle boundary proteins, anti-cdk2, anti-cdk4, anti-cdk6, anti-cyclin D1, anti-cyclin D2, anti-cyclin D3, anti-cyclin E, anti-actin and anti-GAPDH, were purchased from Santa Cruz Biotechnology (Santa Cruz, USA). Anti-p21 and anti-p53 were purchased from Oncogene Science (Cambridge, USA). Anti-p27 was purchased from Calbiochem (Dramstadt, Germany).
Cell-cycle analysis
To measure the cellular DNA content, TREK2 siRNA treated cells were fixed with ethanol in PBS, treated with 20 µg/ml RNase A (Sigma-Aldrich, St Luis, USA) and stained with 50 µg/ml of propidium iodide (Sigma-Aldrich, St Luis, USA). Samples were then analyzed by a fluorescence-activated cell sorting (FACS) on a Calibur flow cytometer (BD Bioscience, San Jose, USA). Modfit LT cell cycle analysis software version 3.0 (Verity software house, Topsham, USA) was used to determine the relative DNA content based on the presence of red fluorescence.
Immunocytochemistry
The 253J cells were fixed with 3.7% paraformaldehyde in phosphate-buffered saline (PBS) for 20 min at room temperature and were permeabilized with 0.5% Triton X-100 in PBS for 3 min. The cells were blocked for 30 min at 25℃ with 3% BSA in PBS. For the staining, the cells were incubated with rabbit polyclonal anti-hKCNK10 (TREK2) (concentration of 1:200, Alomone Labs, Jerusalem, Israel) for overnight at 4℃, followed by incubation with an Alexa Fluor 488 conjugated secondary antibody (concentration of 1:200, Santa Cruz Biotechnology, Santa Cruz, USA) for 1 h. To visualize F-actin, cells were stained with Alexa Fluor 594 conjugated phalloidin (concentration of 1:100, Life Technologies, Grand lsland, USA) for 30 min at 25℃. The images were acquired confocal microscopy (TCS-SP2 AOBS, Leica Microsystems, Heidelberg, Germany).
Statistics
The data are shown as the means±standard error (S. E.), and n represents the number of cells tested. The significance of the differences between the means was established using Student's t-test. p<0.05 was regarded as significant. All statistical analyses were conducted with the Origin program version 6.0 (Microcal, Northampton, USA).
RESULTS
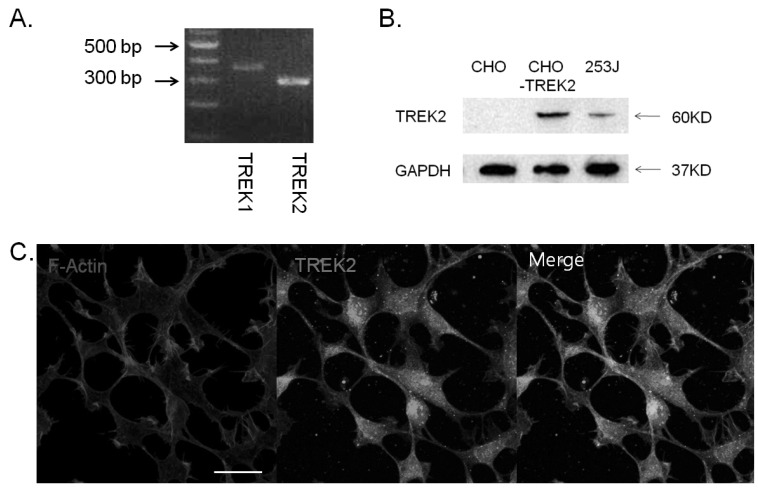
To explore the functional role of TREK2 channels in bladder cancer cells, we first examined their expression in bladder cell lines using qRT-PCR and Western blot (Fig. 1A and Fig. 1B). TREK2 mRNA was expressed and its protein was detected in 253J cells. Figure 1C showed representative confocal microscopic image of TREK2 in 253J cells. TREK2 was observed in plasma membrane and cytosol.
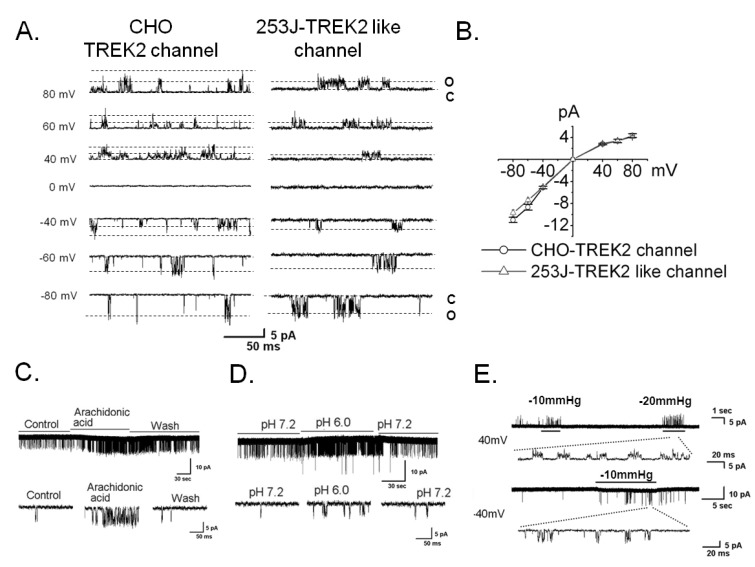
We performed the patch clamp technique to confirm the functional expression of TREK2 using single channel recording in symmetrical 150 mM KCl solutions. We were able to record the TREK2 channel in the bladder cancer cell line 253J but not 5637 or EJ. Thus, we carried out the remainder of the study in 253J cells. The current-voltage (I-V) relation curve showed slightly inward rectification (Fig. 2A and 2B), and the single channel conductance was 123 pS (123.2±5.8 pS, n=7, Fig. 2A) at -60 mV. The channel is similar in TREK2-transiently overexpressing CHO cell, as shown (145.0±8.7 pS, n=4, Fig. 2A). To observe the similarity with the electrophysiological properties of the TREK2 channel, we tested the effect of known physiological stimulants of TREK2 including mechanical stretch, arachidonic acid (AA), and intracellular acid pH (pH 6.0) on the TREK2-like channel in 253J cells (Fig. 2C, 2D, and 2E) [16]. TREK2-like channel activity showed an 11-fold increase due to AA (relative NPo, 10.8±0.6, n=4) and an 8-fold increase due to intracellular acidic pH (relative NPo, 8.5±0.3, n=4) compared with the control (Fig. 2C and 2D). Mechanical stretch also activated the TREK2-like channel at +40 mV and -40 mV in 253J cells (Fig. 2E). These results revealed TREK2 is functionally expressed in bladder cancer 253J cell.
To confirm the effect on membrane potential and cell growth, knockdown was performed with siRNA specific to TREK2. We first measured the level of TREK2 expression on knockdown using qRT-PCR and Western blot 72 hours after transfection. TREK2 mRNA was decreased to ~43% in cells transfected with TREK2 siRNA compared to negative control siRNA (42.9±5.0%, n=8 with TREK2 siRNA, Fig. 3A). The protein level was also decreased by knockdown (Fig. 3B). To measure the membrane potential, we performed current clamp (I=0) with a whole cell patch configuration. The membrane potential was slightly depolarized by knockdown of TREK2 with siRNA (-19.9±0.8 mV, n=116 with negative control siRNA vs. -8.5±1.4 mV, n=74 with TREK2 siRNA) (Fig. 3C). Next, we observed the effect of TREK2 knockdown on cell proliferation. Cell growth decreased to ~64% after TREK2 silencing compared to cells expressing the negative control (63.7±5.2%, n=38 with TREK2 siRNA, Fig. 3D). Figure 3E showed representative images of 253J cells transfected with TREK2 siRNA and negative control siRNA.
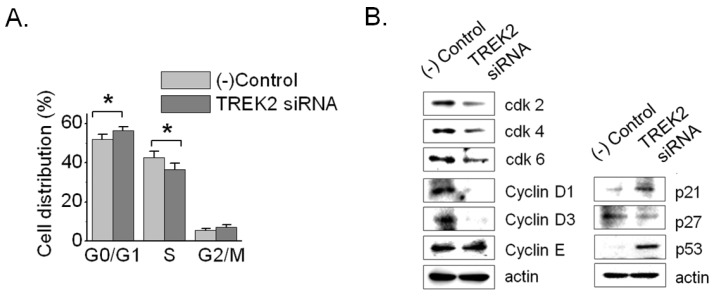
To elucidate the influence of TREK2 knockdown on cell-cycle progression, we checked the DNA content using flow cytometry. The silencing of TREK2 for 72 hours led to 253J cell-cycle arrest, with an increase of cells in the G0/G1 phases (51.8±2.4% in control vs. 56.3±1.9% in TREK2 siRNA, n=8) and a decrease in the percentage of cells in the S phase (42.7±3.3% in control vs. 36.6±3.0% in TREK2 siRNA, n=8) (Fig. 4A). These data prove that the inhibition of DNA synthesis on TREK2 knockdown resulted from cell cycle arrest at G0/G1. Because silencing of TREK2 channels affected G0/G1 phase progression in bladder cancer cells, we examined the expression of central cell cycle regulatory proteins such as cyclin D1, cyclin D3, cdk2, cdk4, cdk6, p21, and p53. The protein levels of p53 and p21 were increased and cyclin D1, cyclin D3, cdk2, cdk4, and cdk6 were decreased (Fig. 4B). Therefore, these results clearly demonstrated that TREK2 was related to G0/G1 phase arrest in bladder cancer growth.
Taken together, TREK2 was functionally expressed in bladder cancer 253J cells and membrane depolarization by knockdown of TREK2 caused cell growth inhibition through cell cycle arrest at the G0/G1 phase.
DISCUSSION
In the present study, we demonstrated for the first time that TREK2 was functionally expressed in bladder cancer 253J cells. Furthermore, membrane depolarization induced by TREK2 knockdown inhibited growth of 253J cells via cell cycle arrest.
Changes in membrane potential by K+ channel regulation are generally accepted to affect the regulation of the proliferative state [1]. Depolarization by K channel blockers leads to increased cell volume by water influx followed by an increase in solute, resulting in decreased proliferation [2,3,17]. Changes in membrane potential may also be involved in the control of cell cycle boundary proteins, such as cyclin-dependent kinase inhibitors and/or cyclin expression [18,19]. Previous reports indicate that the inhibition of KATP or hERG1 (KV11.1) caused cell cycle arrest at the G0/Gl phase [20,21], and cells in G0/G1 phase arrest were depolarized [22-24]. In addition, it has been reported that silencing hEag1 (KV10.1) in cells led to a decrease in cyclin expression and an increase of p21 [25]. These reports support our data that depolarization induced by knockdown of TREK2 with siRNA induced cell growth inhibition and cell-cycle arrest at the G0/G1 phase through an increase of p21 and p53 and a decrease of cyclin D1 and cyclin D3 (Fig. 3 and Fig. 4). Furthermore, the increase in p21 expression inhibited the cyclin D/cdk complex and altered cellular proliferation by regulating cell cycle progression at G1 [26]. In this study, we found that TREK2 knockdown resulted in the downregulation of cyclin D1 and D3, leading us to expect a decrease of cyclin D/cdk complex formation necessary for the G1/S transition (Fig. 4) [26]. In addition, we found an increase in the tumor suppressor gene p53, which delays the G1 phase of the cell cycle [26]. Therefore, these data clearly demonstrated that TREK2 participated in cell cycle arrest at the G0/G1 phase through the down-regulation of cdks kinase activity, via the selective induction of p21-mediated p53 expression.
During the experiments, we needed to distinguish between TREK1 and TREK2 during single channel recording, as both have very similar electrophysiological properties [27-29]. In bladder cancer cells, we were able to distinguish these proteins using single channel conductance and I-V relationships. TREK2 had a higher single channel conductance than TREK1, and TREK2 showed a more inward rectifying I-V relationship than TREK1 in symmetrical 150 mM KCl solutions (Fig. 2). Thus, we could conclude that TREK2 was functionally present in bladder cancer 253J cells because our single channel data were similar to previous results in other cells [28,29]. In present study, we could not record TREK2 channel in 5637 or other cell lines except 253J cell. The reason might be due to TREK2 translocation deficiency into plasma membrane by unknown signals or a low density of the channels in plasma membrane.
TREK2 generally has been known to be activated by ischemic insults such as osmotic stress or change in pH to regulate resting membrane potential. Consequently, the TREK2 channel might act to protect the cancerous cells, similar to TASK3, which promotes tumor formation [7]. Therefore, the physiological relevance of the TREK2 channel requires further study to provide a candidate for adjuvant therapy along with anticancer drugs, as well as to provide the mechanism of disease progression, recurrence, and therapy in bladder cancer.

 XML Download
XML Download