

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
DHEA
dehydroepiandrosterone
HSD
hydroxysteroid dehydrogenase
NMDA
N-methyl-D-aspartate receptor
THDOC
allotetrahydrodeoxy corticosterone
DHT
dihydrotestosterone
T
testosterone
CBZ
carbamazepine
TTC
triphenyltetrazolium chloride
NAD
nicotinamide adenine dinucleotide
TBARS
thiobarbituric acid reactive substances
SEM
standard error mean
TLT
transfer latency time
Indo
indomethacin
Mexil
mexiletine
GABA
gamma-aminobutyric acid
MDA
malondialdehyde
NAD
nicotinamide adenine dinucleotide
NADPH
nicotinamide adenine dinucleotide phosphate
CPCSEA
committee for the purpose of control and supervision of experiments on animals
INTRODUCTION
Stroke is a syndrome characterized by rapid onset of neurological injury due to interruption of blood flow to the brain. Although mortality from stroke has declined over the last decade but it still remains the third leading cause of death after heart attack and cancer and is a major cause of morbidity particularly in middle aged and elderly population. The majority of strokes result from embolic or thrombotic obstruction of cerebral vasculature. Stroke leads to ischaemic brain injury. Moreover, stroke is associated with higher incidence of deficits in sensory motor functions and cognitive ability [1,2].
The brain is capable of de novo biosynthesis of neurosteroids independent of gonads, adrenals, placenta and other peripheral steroidogenic organs [3,4]. The most active steroidogenic cells in the brain include astrocytes, oligodendrocytes and neurons. Synthesis of neurosteroids involves a series of enzymatic reactions mediated by P450 enzymes like P450scc, P450c17, P450 arom and non-P450 enzymes like 17α-HSD, 3-αHSD, 3α-HSD, 5α-reductase etc [5].
The major neuroactive steroids include allopregnanolone, dehydroepiandrosterone (DHEA), dehydroepiandrosterone sulphate (DHEAS), pregnenolone, pregnenolone sulphate, progesterone and allotetrahydrodeoxycorticosterone (THDOC). The prevention of over activation of NMDA receptors by neurosteroids appears to be useful in stroke and other neurodegenerative diseases [6,7]. DHEA has shown to prevent oxidative brain injury [8]. Allopregnanolone reduces glutamate-induced irreversible changes in intracellular Ca2+ concentrations and provides neuroprotection through GABA receptors [9]. The functional and morphological neuroprotective effects of androgens like Testosterone (T), dihydrotestosterone (DHT), 5 alpha-androstane-3 alpha, 17 beta-diol (3α DIOL) are mediated through neurosteroids [10].
The enzyme 3α-HSD catalyzes the reversible reduction of androgen DHT to 3α DIOL. It showed higher activity with the phosphorylated cofactors, NADPH and NADP+ and was inhibited by indomethacin [11]. The enzyme 3α-HSD also catalyzes allopregnanolone biosynthesis from its immediate precursor 5α-dihydroprogesterone [12]. Antidepressants (desipramine, amitriptyline), anticonvulsants (phenytoin, diazepam, carbamazepine) and addictive drugs (amphetamine, morphine) increase allopregnanolone synthesis by stimulating the activity of the enzyme, 3α-HSD, in the frontal cortex [13]. Selective Serotonin Reuptake Inhibitors (SSRIs) like fluoxetine, paroxetine and sertraline also increase the efficiency of the enzyme 3α-HSD resulting in dramatically enhanced allopregnanolone formation. The studies support the possibility that SSRI-induced allopregnanolone biosynthesis may be relevant to SSRI therapeutic actions in depression and anxiety [14].
Carbamazepine (CBZ) inhibits voltage-dependent sodium channels and is used for treating epilepsy and trigeminal neuralgia [15]. Administration of CBZ to rats increases the concentrations of pregnenolone, progesterone, allopregnanolone, and allotetrahydrodeoxycorticosterone in both the cerebral cortex and plasma by stimulating the activity of the enzyme, 3α-HSD, in the frontal cortex, and these neurosteroids may be partly involved in the mechanism of action of this drug [13]. CBZ is also noted to induce steroidogenesis in the brain of adrenalectomized-orchiectomized rats [16]. CBZ regulates neuronal excitability by stimulating pregnenolone biosynthesis which involve a direct or indirect interaction with peripheral type benzodiazepine receptors [17]. Indomethacin is a non-selective inhibitor of cyclooxygenase (COX) 1 and 2, enzymes that participate in prostaglandin synthesis from arachidonic acid. Indomethacin is also a known inhibitor of the enzyme 3α-HSD [18]. Mexiletine is a member of the class Ib antiarrhythmic drugs. It primarily acts by blocking fast sodium channels thereby reducing the phase 0 maximal upstroke velocity of the action potential [19]. Based on the above, the present study was designed to investigate the effects of carbamazepine in ischaemia-reperfusion-induced cerebral injury and to observe the modulatory effects of indomethacin and mexiletine on carbamazepine mediated actions.
Go to : 
METHODS
Experimental animals
Swiss albino mice of either sex weighing 20~30 g maintained on a standard laboratory diet (Kissan Feeds Ltd., Mumbai, India) and tap water ad libitum were employed in the present study. They were housed in the departmental animal house and were exposed to natural photoperiod. The experiments were conducted in a semi-sound proof laboratory between 10:00 am to 5:00 pm. The protocol of the study was approved by the institutional animal ethics committee and the care of the animals was carried out according to the guidelines of the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Ministry of Environment and Forest, Government of India (Reg. No.-107/1999/CPCSEA).
Drugs and chemicals
Indomethacin (Sun Pharmaceutical Ltd., Mumbai, India) was dissolved in sodium carbonate solution. Carbamazepine (Torrent Laboratories Pvt. Ltd., Ahmedabad, India) was dissolved in 25% ethyl alcohol solution. Chloral Hydrate (400 mg/kg, i.p.; Reidal-deHaen, Germany) and Mexiletine (German Remedies Ltd., Mumbai, India) were dissolved in water. All other chemicals were of analytical quality. All drug solutions were freshly prepared before use. The dose selection is made on the basis of previous reports [20] and our pilot studies.
Global cerebral ischaemia and reperfusion
Mice were anesthetized with chloral hydrate (400 mg/kg, i.p.). A midline ventral incision was made in the throat. Right and left common carotid arteries were located and freed from surrounding tissue and vagus nerve. A cotton thread was passed below each of the carotid artery. Global cerebral ischaemia was induced by pulling the ends of thread with constant weight. After 10 min of global cerebral ischaemia, weight on the thread was removed to allow the reflow of blood through carotid arteries. The incision was sutured back in layers. The sutured area was cleaned with 70% ethanol and was sprayed with antibiotic (neosporin) dusting powder so as to prevent any infection. Body temperature of mice was maintained at 37℃ during the surgery by electrically heated surgical platform during reperfusion of 24 h by housing the animal cage inappropriate temperature and humidity-controlled room [21]. After completion of the surgical procedure, the animals were shifted individually to their home cage and were allowed to recover.
Assessment of cerebral infarct size
At the end of 24-h reperfusion after the global cerebral ischaemia, animals were sacrificed by cervical dislocation and the brain was removed. The brain was kept overnight at -4℃. Frozen brain was sliced into uniform coronal sections of about 1mm thickness. The slices were incubated in 1% triphenyltetrazolium chloride (TTC) at 37℃ in 0.2 M Tris buffer (pH 7.4) for 20 minutes [22,23]. TTC is converted to red formazone pigment by nicotinamide adenine dinucleotide (NAD) and dehydrogenase and thereof stained the viable cells deep red. The Infarcted cells have lost the enzyme and cofactor and thus remained unstained dull yellow. The brain slices were placed between two glass plates and a transparent plastic grid with 100 squares in 1 cm2 was placed above it. The average area of the brain slice was calculated by counting the number of squares on either side. Similarly, numbers of square falling over non-stained dull yellow area were counted. Infarct size was expressed as percentage of total brain volume. The whole of the brain slices were weighed, the infarcted dull yellow part was dissected out and weighed separately. The infarct size was expressed as a percentage of total wet weight of brain.
Estimation of thiobarbituric acid reactive substance (TBARS)
At the end of 24 h of reperfusion after the global cerebral ischemia, animals were sacrificed by cervical dislocation and the brain was removed. The brain was homogenized in 5 ml of 30 mM Tris-HCl and 2.5 mM CaCl2 buffer (pH 7.6 at 5℃). Homogenate was centrifuged at 750 g to separate cellular debris. The supernatant was accurately divided into two parts. Both portions were centrifuged at 8,200 g to obtain the mitochondrial fraction. One fraction (both mitochondrial pellet and supernatant) was utilized for determination of TBARS [24] and the other portion (both mitochondrial pellet and supernatant) was employed in protein estimation by Lowry's method [25]. The extent of lipid peroxidation was expressed as nanomoles of TBARS formed per mg of protein.
Inclined beam walking test
Inclined beam-walking test was employed to evaluate fore and hind limb motor co-ordination [26]. Each animal was individually placed on a metallic bar 55 cm long and 1.5 cm wide, inclined at an angle of 60° from the ground. The motor performance of mice was graded on a scale ranging from 0 to 4. A grade of 0 was assigned to an animal that could readily traverse the beam, grade 1 was given to animal demonstrating mild impairment, grade 2 was assigned to animal demonstrating moderate impairment, grade 3 was given to animal demonstrating severe impairment and grade 4 was assigned to animal completely unable to walk on the beam. Inclined beam-walking test was performed before global cerebral ischaemia and the animals which readily traverse the beam (grade 0) were selected. The test was again performed 24 h after global cerebral ischaemia and reperfusion.
Rota rod test
Rota rod is used to evaluate motor coordination by testing the ability of mice to remain on a revolving rod [27]. The rate of rotation of the rod was adjusted such that the normal mouse was able to stay on the rotating rod for a period of five minutes. The difference in the fall off time from the rotating rod between the control and treated animals is taken as an index of motor in coordination. The mice that are able to stay on the rotating rod for a period of five minutes before global cerebral ischaemia were selected and the test was again performed 24 h after global cerebral ischaemia and reperfusion.
Experimental design
Animals were randomly separated into eight groups and each group comprised of 10 mice. The sequence of tests performed on each animal was Rota-rod test, beam walking test and elevated plus-maze test. The gap of 10 minutes was kept between each test. These tests were performed in the above described sequence 3 h before global cerebral ischaemia and 24 h after global cerebral ischaemia and reperfusion.
1. Sham group (group I)
Mice were subjected to surgical procedure under chloral hydrate anesthesia (400 mg/kg, i.p.) and a thread was passed below both carotid arteries, but the arteries were not occluded. After 10 min, the thread was removed and the animal was sutured back and allowed to recover for 24 h.
2. Control group (group II)
Mice were subjected to 10 min global cerebral ischaemia followed by reperfusion for 24 h.
3. Vehicle treated group (group III)
Mice were administered vehicle (25% ethyl alcohol, 0.28 ml) 1 h before global cerebral ischaemia of 10 min followed by reperfusion for 24 h.
4. Carbamazepine treated group (group IV)
Mice were administered carbamazepine (16 mg/kg, i.p.) 1 h before global cerebral ischaemia of 10 min followed by reperfusion for 24 h.
5. Indomethacin treated group (group V)
Mice were administered indomethacin (100 mg/kg, i.p.) 2 h before global cerebral ischaemia of 10 min followed by reperfusion for 24 h.
6. Indomethacin and carbamazepine treated group (group VI)
Mice were administered indomethacin (100 mg/kg, i.p.) 2 h before, and carbamazepine (16 mg/kg, i.p.) 1 h before global cerebral ischaemia of 10 min followed by reperfusion for 24 h.
Statistical analysis
The results were expressed as mean±standard error of means (S.E.M.). Statistical analysis for transfer latency time (TLT), thiobarbituric acid reactive substances, infarct size and rota rod test was performed using one way analysis of variance (ANOVA) followed by Tukey's multiple range tests as post-hoc analysis for multiple comparisons. Statistical analysis for inclined beam walking test was performed using the Wilcoxon rank sum test. A value of p<0.05 was considered to be statistically significant.
Go to :
RESULTS
Effect of various interventions on cerebral infarct size
Global cerebral ischaemia of 10 min. followed by reperfusion for 24 h produced a significant increase in cerebral infarct size measured by volume and weight method. The administration of vehicle (25% ethyl alcohol, i.p.) 1 h before induction of cerebral ischaemia did not affect ischaemia-reperfusion-induced increase in cerebral infarct size. Carbamazepine (16 mg/kg, i.p.), a stimulator of 3α-hydroxysteroid dehydrogenase, administered 1h before the induction of cerebral ischaemia significantly attenuated ischaemia-reperfusion-induced increase in cerebral infarct size. Pretreatment of indomethacin (100 mg/kg, i.p.), an inhibitor of 3α-hydroxysteroid dehydrogenase, attenuated the effect of carbamazepine on cerebral infarct size. However, no per se effect of indomethacin was observed on infarct size. Administration of mexiletine (50 mg/kg, i.p.), a sodium channel blocker, produced a slight decrease in infarct size but results were not statistically significant. Similarly, Pretreatment of indomethacin did not modulate the effect of mexiletine on ischaemia-reperfusion-induced cerebral infarct size (Fig. 1).
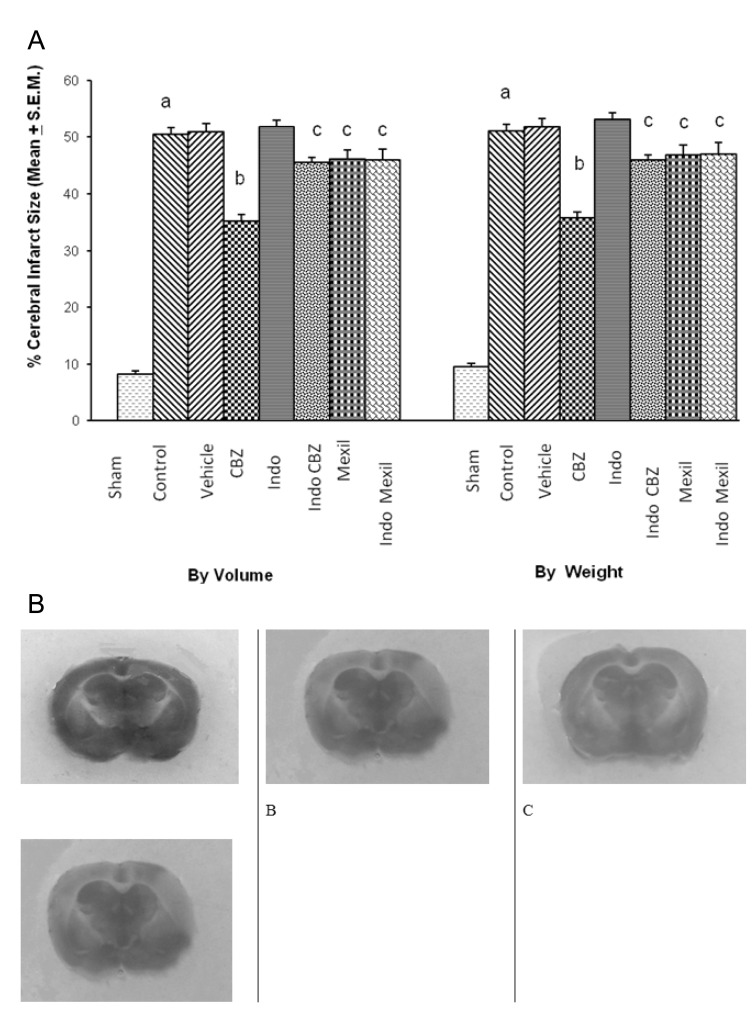 | Fig. 1(A) Effect of carbamazepine on ischemia reperfusion-induced cerebral infarct size and its modulation by mexiletine and indomethacin. CBZ, carbamazepine; Indo, indomethacin; Mexil, mexiletine. Values are expressed as mean (n=10)±S.E.M. a=p<0.05 vs sham; b=p<0.05 vs control; c=p<0.05 vs carbamazepine. (B) The representative photographs of TTC staining showing cerebral infarcted (yellow colour) and non-infarcted (red colour) areas. A, sham group; B, ischemia-reperfusion injury group; C, carbamazepine treated group; D, indomethacin+carbamazepine treated group.
|
Effect of various interventions on Thiobarbituric Acid Reactive Substances (TBARS)
Global cerebral ischaemia of 10 min followed by reperfusion of 24 h significantly increased TBARS in mitochondrial and supernatant fraction. Carbamazepine significantly attenuated ischaemia-reperfusion-induced increase in TBARS. Pre-treatment with Indomethacin attenuated the effect of carbamazepine on ischaemia-reperfusion-induced TBARS. However, no per se effect of indomethacin was observed on ischaemia-reperfusion-induced TBARS. Administration of mexiletine produced a slight decrease in ischaemia-reperfusion-induced TBARS but the results were not statistically significant. Pre-treatment of indomethacin did not modulate the effect of mexiletine on ischaemia reperfusion induced increase in TBARS (Fig. 2).
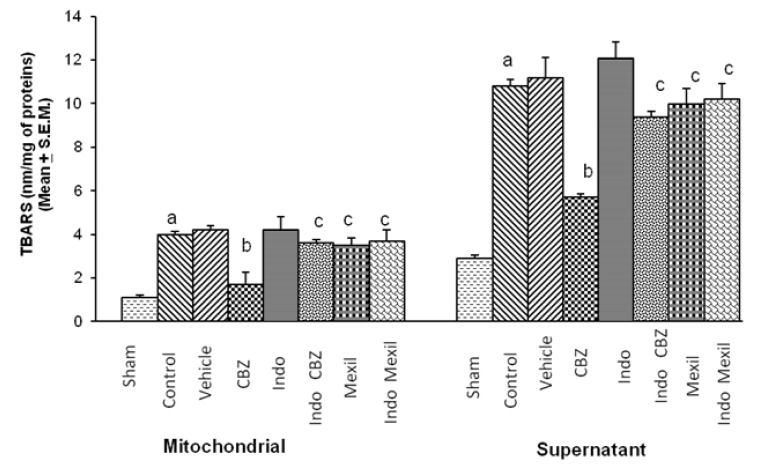 | Fig. 2Effect of carbamazepine on ischemia reperfusion-induced changes in thiobarbituric acid reactive substances levels in the brain (TBARS) and its modulation by mexiletine and indomethacin. CBZ, carbamazepine; Indo, indomethacin; Mexil, mexiletine. Values are expressed as mean (n=10)±S.E.M. a=p<0.05 vs sham; b=p<0.05 vs control; c=p<0.05 vs carbamazepine.
|
Effect of various interventions on motor co-ordination
Global cerebral ischaemia of 10 min. followed by reperfusion for 24 h produced a marked impairment of motor performance using inclined beam walking and rota-rod test (Fig. 3 and 4). Carbamazepine administered 1 h before the induction of cerebral ischaemia significantly attenuated ischaemia-reperfusion-induced impairment of motor performance. Pre treatment with Indomethacin attenuated carbamazepine induced improvement of motor performance. However, no per se effect of indomethacin was observed on ischaemia-reperfusion-induced impairment of motor performance. Mexiletine improved ischaemia-reperfusion-induced impairment of motor performance, however, the results were not statistically significant. Also, pretreatment with Indomethacin did not modulate the effect of mexiletine. Moreover, the improvement of motor performance by indomethacin and mexiletine was not statistically significant as compared to control. The motor performance assessed by inclined beam walking and rota-rod tests demonstrated identical results with all treatments (Fig. 3 and 4).
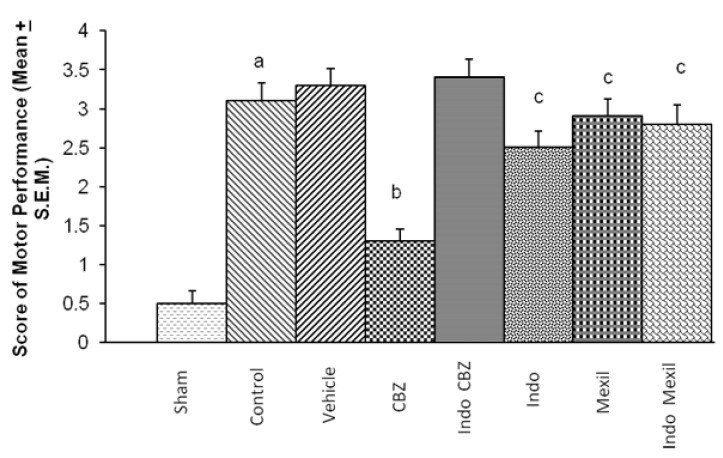 | Fig. 3Effect of carbamazepine on ischemia reperfusion-induced impairment on motor performance and its modulation by mexiletine and indomethacin. CBZ, carbamazepine; Indo, indomethacin; Mexil, mexiletine. Values are expressed as mean (n=10)±S.E.M. a=p<0.05 vs sham; b=p<0.05 vs control; c=p<0.05 vs carbamazepine.
|
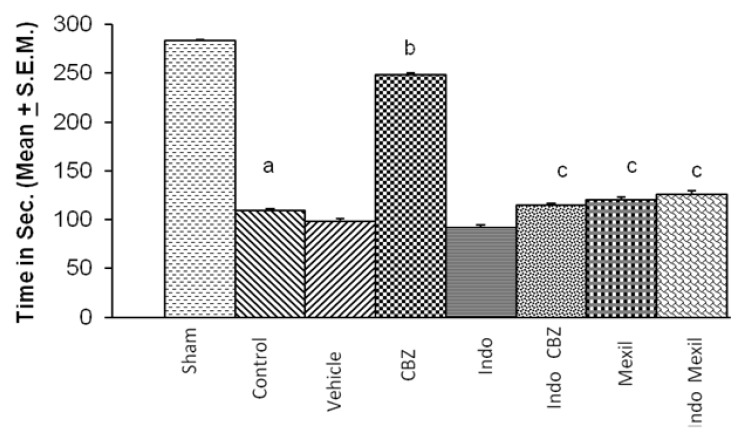
Go to :
DISCUSSION
The results of the present study indicate that global cerebral ischemia and reperfusion produced significant neuronal injury manifested in the terms of an increase in infarct size, motor incoordination and oxidative stress (indicated by a rise in TBARS levels). Above effects of global cerebral ischemia/reperfusion were significantly abolished by pretreatment of carmabazepine.
The noted neuronal injury (increase in infarct size & motor incoordination) induced by global cerebral ischemia/reperfusion is in line with our earlier reports [21,26] and reports from other laboratories [28,29].
In our study, Global cerebral ischemia/reperfusion induced neuronal injury was significantly attenuated by pretreatment of Carbamazepine. Carbamazepine is noted to stabilize the inactivated state of sodium channels making neurons less excitable [30]. Carbamazepine has also been reported to increase the concentrations of allopregnanolone, pregnenolone, progesterone and THDOC in the cerebral cortex as well as in plasma [16], perhaps by stimulating the enzyme 3α-hydroxysteroid dehydrogenase (3α-HSD) [13,16]. The allopregnanolone and pregnenolone has been documented to produce a neuroprotective effect [31,32] by activating GABA receptors [33] and by inhibiting glutamate induced irreversible changes in the intracellular Ca2+ concentration [34]. Therefore, it may be possible to speculate that carbamazepine induced attenuation of ischaemia-reperfusion-induced increase in cerebral infarct size noted in the present study is due to increase in synthesis of neurosteroids. This contention is further supported by the observation of the study at hand that indomethacin, an inhibitor of 3α-HSD [18,35] has attenuated the neuroprotective effect of carbamazepine. Carbamazepine has been reported to produce blockade of sodium channels [30] which may also be contributing to its neuroprotective effect. On the other hand, mexiletine, a sodium channel blocker, has shown a tendency to decrease ischaemia-reperfusion-induced rise in cerebral infarct size but without any statistical significance. Moreover, mexiletine has not modulated the effect of indomethacin on ischaemia-reperfusioninduced increase in cerebral infarct size. It indirectly suggests that sodium channel blocking effect of carbamazepine may not be contributing markedly towards its neuroprotective effect. Mexiletine has been reported to exert neuroprotective effects at higher doses [36] than that we employed in this study, however, it has also been reported to block sodium channels at low doses [20]. We tried to use mexiletine exclusively for its sodium channel blocking action and not for its neuroprotective effect.
The depletion of ATP due to ischaemia leads to cellular polarization and activation of NMDA receptors through the release of glutamate from presynaptic nerve terminals which consequently increase the influx of Ca2+ inside the cell [37]. The increased cytosolic Ca2+ activates calpain, calcineurin, phospholipase A2 and phospholipase C [38] which generate free radicals. Reperfusion is also documented to generate superoxide anions from arachidonic acid metabolism and promotes nitric oxide formation by activation of nitric oxide synthase [39]. These free radicals are implicated in cerebral ischaemia and reperfusion-induced neuronal injury [40,41]. Free radicals promote lipid peroxidation which results in the alteration in permeability and fluidity of membranes [42]. The neurons vulnerable to free radical damage during reperfusion are reported to demonstrate lipid peroxidation. The concentration of malondialdehyde (MDA), which is an end product of lipid peroxidation, increases markedly after reperfusion of the brain [43]. Thus in the present study the noted increase of TBARS in mitochondrial and supernatant fractions of brain tissue as a result of ischaemia and reperfusion may be due to the generation of free radicals. Carbamazepine has attenuated ischaemia-reperfusion-induced increase in TBARS in mitochondria and cytoplasm, which is the index of lipid peroxidation. This may be due to the fact that carbamazepine has increased the synthesis of neurosteroids which in turn may have reduced glutamate induced Ca2+ overload [34] and hence has decreased lipid peroxidation and TBARS. This is further supported by the observation that indomethacin, an inhibitor of 3α-HSD, has attenuated carbamazepine induced decrease in TBARS. Moreover the noted effect of carbamazepine on TBARS may not be due to its effect on sodium channels because mexiletine, a sodium channel blocker, has not modulated the effect of carbamazepine on TBARS in mitochondria and cytoplasm. So it may be concluded that the effect of carbamazepine on TBARS may be due to increase in synthesis of neurosteroids.
Brain tissue infarction as a result of oxygen free radicals has been noted to decrease motor performance and motor co-ordination. The increase in neurosteroid synthesis due to activation of 3α-HSD and the consequent prevention of generation of reactive oxygen species [34] may be responsible for the noted ameliorative effect of carbamazepine on ischaemia-reperfusion-induced impairment of motor performance. This point is further supported by our observation with indomethacin, an inhibitor of 3α-HSD, who attenuated the neuroprotective effect of carbamazepine on motor performance. Further, the ameliorative effect of carbamazepine on motor performance does not seem to involve its sodium channel blocking property because mexiletine, a sodium channel blocker, has not improved ischaemia-reperfusion-induced impairment of motor performance.
Therefore, it appears evident from our results that carbamazepine is showing its neuroprotective effects probably by increasing neurosteroid synthesis as a consequence of 3α-hydroxysteroid dehydrogenase enzyme activation and not through its sodium channel blocking action. However, further studies are needed to clear some points like any possible role of COX inhibitory action of indomethacin in its attenuation of carbamazepine effect and any contribution of difference in molecular identity between central and peripheral sodium channels to our findings.
With data in hand, it may be concluded that the neuroprotective effect of carbamazepine may be due to increase in synthesis of neurosteroids perhaps through activation of enzyme 3α-hydroxysteroid dehydrogenase and its sodium channel blocking effect is probably not implicated in its neuroprotective effect. Nevertheless further studies are needed to substantiate these findings.
Go to :
 XML Download
XML Download