

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
ATRA
all-trans retinoic acid
CRABP
cellular retinoic acid-binding protein
CRBP
cellular retinol-binding protein
CYP26A1
cytochrome p450 26A1
eWAT
epididymal white adipose tissue
FABP5
fatty acid binding protein 5
HBV
hepatitis B virus
HBx
HBV x antigen
HCC
hepatocellular carcinoma
LRAT
lecithin:retinol acyltransferase
NAFLD
non-alcoholic fatty liver disease
PPAR
peroxisome proliferation-activated receptor
RAR
retinoic acid receptor
RXR
retinoid X receptor
Tg
transgenic
Tgm2
tissue transglutaminase 2
INTRODUCTION
Hepatitis B virus (HBV) infection has been shown to be associated with the increase in the risk of type 2 diabetes [1]. HBV x antigen (HBx) enhances lipogenesis and gluconeogenesis in liver of whole body HBx-transgenic (HBx Tg) mice, leading to hepatic steatosis and impaired glucose tolerance (IGT) [2,3]. HBx gene is the smallest of the four partially overlapping open reading frames of HBV [4]. Because HBx is involved in hepatic carcinogenesis [2,5] and because both non-alcoholic fatty liver disease (NAFLD) and diabetes are associated with the increased risk of HCC [6], we have tried to find out any connection between these factors. When we analyzed the expression of various genes in the liver from HBx Tg mice, among prominent differences in gene expression between HBx Tg mice and wild type (WT) control mice was cellular retinol-binding protein-I (CRBP-I) gene.
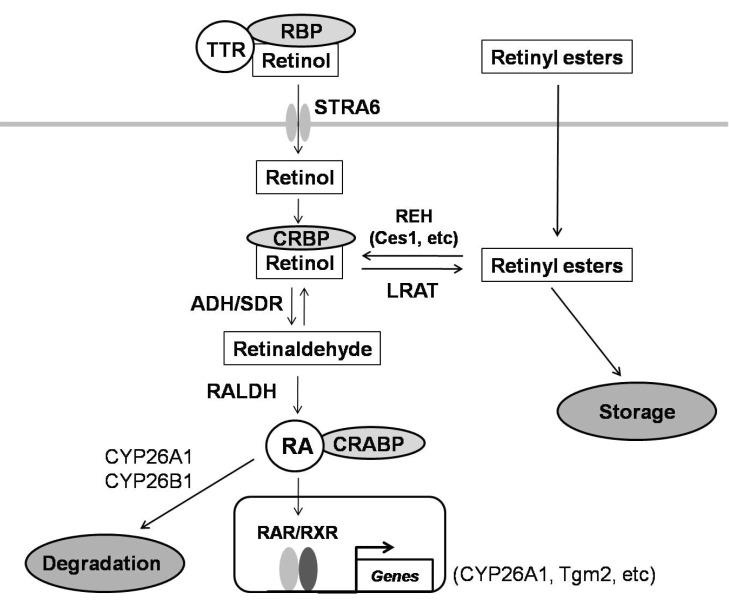
Due to their roles in many cellular processes including differentiation and proliferation, vitamin A metabolites, known as retinoids, have received considerable attentions [7-9]. Retinoic acid (RA), especially all-trans RA (ATRA), is main signaling retinoid in the body [10]. In the cells, retinoids are bound to specific binding proteins. All-trans-retinol and its oxidation product, all-trans-retinal, are associated with one of the two cellular retinol-binding proteins (CRBP-I and CRBP-II), with most intracellular retinol binding to CRBP-I. ATRA binds to one of the two cellular retinoic acid-binding proteins (CRABP-I and CRABP-II). CRBP-I facilitates esterification of retinol in stellate cells by lecithin:retinol acyltransferase (LRAT) to produce retinyl esters for storage, indicating its important role in the regulation of intracellular retinoid storage and metabolism [11]. CRBP-I, specifically expressed in preadipocytes, suppresses adipocyte differentiation, controlling adiposity in vivo [12]. In addition, the hepatic expression of CRBP-I was shown to be significantly higher in patients with NAFLD than in normal subjects [13]. Moreover, genetic manipulation of various retinoid-metabolizing enzymes and retinoid-binding proteins results in alterations in adiposity of mice [14,15]. Our observation of very high expression of CRBP-I in the liver of HBx Tg mice prompted us to evaluate the association of the change in CRBP-I expression with any characteristic change in the retinoid pathway in the liver of HBx Tg mice. Because CRBP-I and retinoid pathway are related with lipid homeostasis and adiposity, we also evaluated the expression levels of CRBP-I in other animal models of insulin resistance (IR). In addition, we assessed the effect of chronic metformin treatment, an anti-diabetic agent that has been shown to improve NAFLD [13] and decrease the incidence of HCC in patients with diabetes [16,17], on hepatic CRBP-I expression and the development of HCC in HBx Tg mice.
Go to : 
METHODS
Animal models
HBxTg mice that have one copy of HBx transgene in chromosome 2 were available from professor Yu DY's laboratory and the details about the development and the characteristics of HBx Tg mice has been described previously [2,5]. Most of experiments and analyses were done in wild type C57BL/6J male mice (WT) and HBx Tg male mice on normal chow diet at their 4 months of age if unspecified. In addition, to determine the effect of metabolic syndrome and fatty liver on CRBP-I expression, WT and HBx Tg mice were placed on a high-fat (HF) diet (D12492, 60% calories from fat; Research Diets, New Brunswick, NJ) from 5 weeks of age for 18 weeks. At the end of the dietary regimen, all mice were sacrificed after an overnight fasting. Blood and tissues, including liver and epididymal white adipose tissue (eWAT), were taken. The tissues and serum were stored at -80℃ until analysis. In addition, male leptin receptor-deficient (db/db) mice (The Jackson Laboratory) of similar age were included in experimental groups for comparisons.
A subgroup of chow-fed HBx Tg mice was treated with metformin (250 mg/kg daily in drinking water) from 6 weeks of age till 18 months of age to determine if metformin has a anti-tumor effect in this mouse model.
All animal experiments were carried out in accordance with the National Institutes of Health's Guide for the Care and Use of Laboratory Animals and were approved by the Institutional Animal Care and Use Committee of Jeju National University.
Western blot analysis
The frozen tissues were homogenized in cold lysis buffer. Aliquots of 60 µg protein from tissue homogenate were subjected to standard western blotting as previously described [18]. The band intensities of the proteins were analyzed by using National Institutes of Health Image J 1.42 software, and the levels of phosphorylated proteins were normalized by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) protein level or its total protein amounts.
RNA Preparation and Real-time quantitative PCR(QPCR) analysis
Liver from WT and HBx Tg mice (4 mice each) were excised in RNAse-free medium, transferred immediately into extraction reagent (TRIzol; Invitrogen, Rockville, MD) on ice. Total RNA was isolation and QPCR were performed as previously described [18]. The genes involved in retinoid physiology as shown in Fig. 6 were analyzed and normalized to the level of GAPDH: Retinol-binding protein 4 (RBP4), stimulated by retinoic acid gene 6 (STRA6), alcohol dehydrogenase (ADH), short-chain dehydrogenase/reductase (SDR), retinyl ester hydrolase (REH), CRBP-I, CRABP-II, LRAT, retinaldehyde dehydrogenases (RALDH1, 2, 3, 4), retinoic acid receptors (RARα, β, γ), retinoid X receptors (RXRα, β, γ), fatty acid binding protein 5 (FABP5), tissue transglutaminase 2 (Tgm2), cytochrome P450 26A1 (CYP26A1), and peroxisome proliferation-activated receptor-α, -δ, and -γ (PPARα, δ, γ). Primer sequences are available upon request.
Histological examination
The livers were isolated from the experimental mice and fixed in 10% formaldehyde. The fixed tissues were embedded in paraffin and were sectioned (4.0 µm). The sections were stained with hematoxylin-eosin and were examined under light microscope to determine the presence or absence of HCC. The development of small neoplastic nodules and grossly identified HCCs were determined at the age of around 18 months as described previously [5]. The pathologist who examined the samples was blinded to the experimental groups.
Retinol and retinoic acid extraction and HPLC analysis
For the extraction of retinoids from the liver of WT and HBx Tg mice, the procedures that Ribaya-Mercado et al. [19] have described were used with some modifications. The HPLC system consisted of a Series 515 LC pump, an 2998 photodiode array detector, and an 2707 autosampler (Waters., Miford, MA, USA) was used for retinol and RA analyses. The reversed-phase system included a Sunfire™ C-18 (5 µm; 4.6×250 mm) ODS column (Waters., Miford, MA, USA). Retinol and ATRA were detected at 325 nm and 350 nm, respectively. The conditions for retinol analysis were minor modifications of the procedure that was reported by Green et al. [20]. The conditions for ATRA analyses were a modification of the procedure that was performed by Tang and Russell [21]. Peak areas were calculated using Waters Empower 3 software (Waters., Miford, MA, USA), and retinol and ATRA mass was determined by an internal standard method, using the mass-to-area ratios for the retinol and ATRA standard curves. The values were presented as µg/gram of dry lipid extract.
Statistical analysis
All results were presented as mean±SEM. For determining statistical significance, a Student's t test or one-way ANOVA with Turkey's post hoc analysis was performed using (SPSS program, version 12.0.0; SPSS, Chicago, IL), whereas Fisher's exact test was performed to compare of the incidence rate of HCC among the experimental groups.
Go to :
RESULTS
The changes in the expression of genes involved in hepatic retinoid pathway including CRBP-I in HBx Tg mice
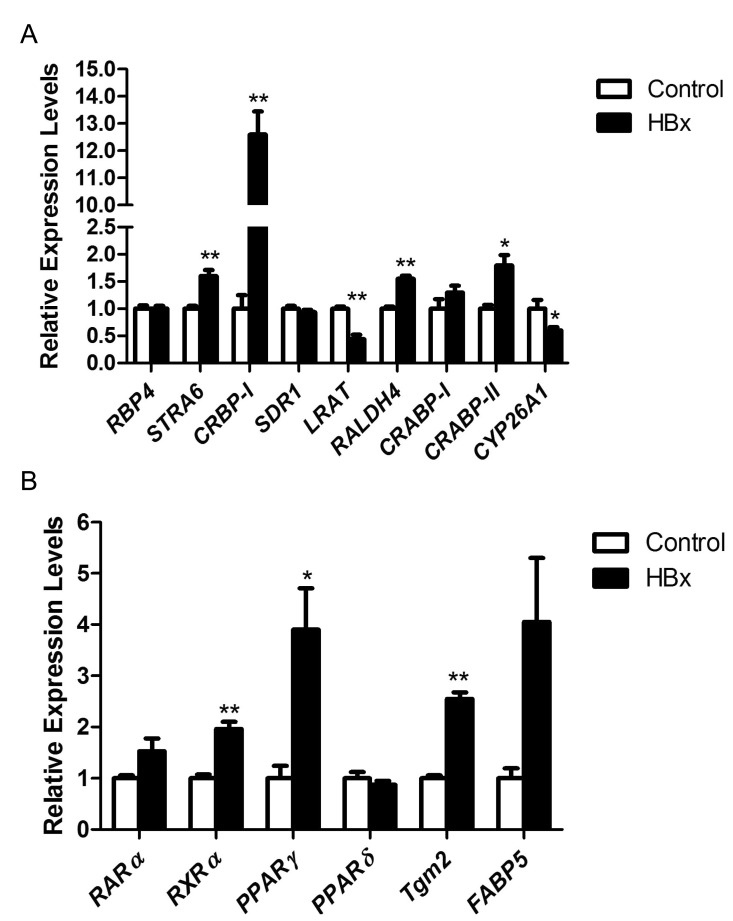
Among the genes involved in the retinoid metabolism, STRA6, CRBP-I, RALDH4, and CRABP-II were up-regulated in the liver of HBx Tg mice compared with WT control mice. In the liver of HBx Tg mice, LRAT was down-regulated (Fig. 1A). We assessed the expression levels of genes involved in retinoid signaling pathway and the target genes of RA signaling. Among the genes for RARs and RXRs, only RXRα was significantly up-regulated. Of the two RA target genes (CYP26A1 and Tgm2) that we tested, Tgm2 was significantly up-regulated whereas CYP26A1 was down-regulated. RA carried into the nucleus by FABP5 acts as PPARγ ligand, whereas RA carried by CRABP-II acts on RAR [22]. In these regards, CRABP-II was significantly up-regulated in HBx Tg mice compared with WT mice, with strong tendency toward an increase in the expression of FABP5 (p=0.053). Hepatic PPARγ expression was significantly increased in HBx Tg mice compared with WT mice (Fig. 1B).
The increased expression of CRBP-I protein and tissue retinoid levels in liver of HBx Tg mice
We assessed CRBP-I protein levels in eWAT and liver of WT control mice and HF-fed control mice, db/db mice, and HBx Tg mice. As shown in Fig. 2, hepatic CRBP-I protein was markedly increased in HBx Tg mice compared with WT control mice. However, hepatic CRBP-I expression in HF-fed mice and db/db mice was comparable to that of WT control mice. HF feeding did not further increase hepatic CRBP-I protein level in HBx Tg mice (data not shown). CRBP-I protein levels in eWAT were comparable between the experimental groups.
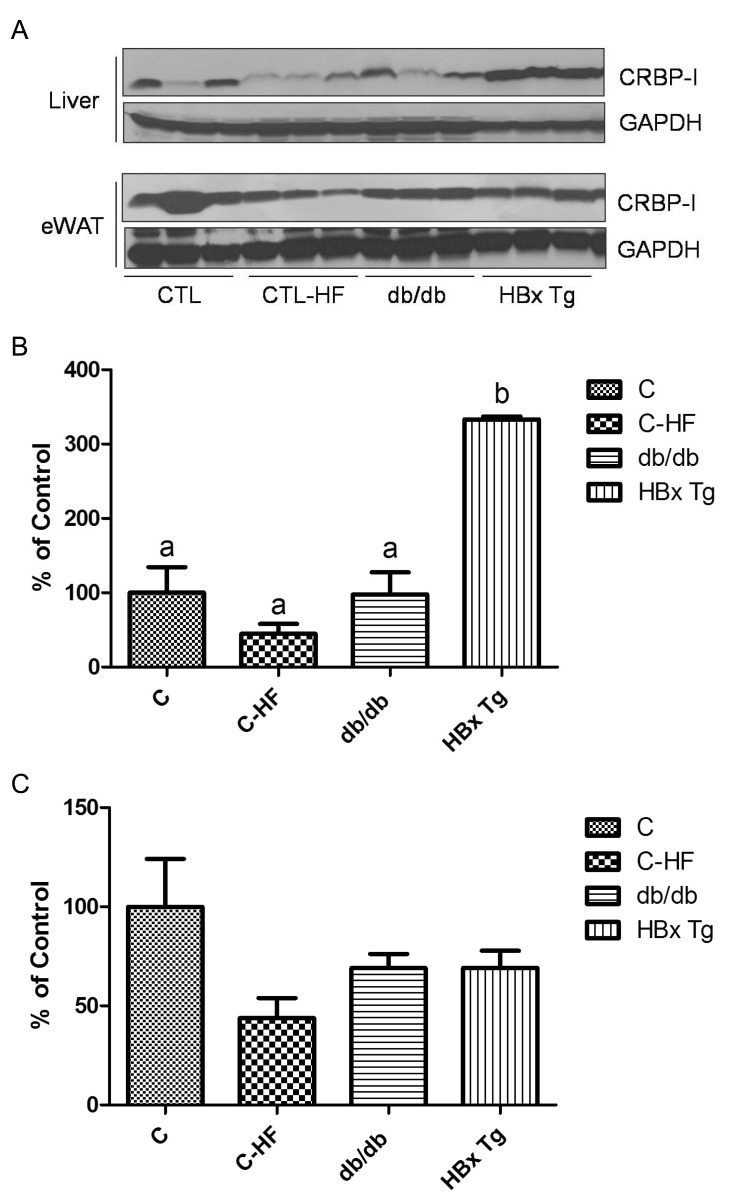 | Fig. 2CRBP-I protein expression in liver and eWAT of WT control mice (C), HF-fed control mice (C-HF), db/db mice, and HBx Tg mice (n=6 mice in each group). (A) The representative immunoblotting images. (B, C) The levels of protein expression presented as the percentage of the mean expression level of the WT control group (% of control). Alphabetical letters on the bars indicated statistical differences (p<0.05) among the experimental groups: the same letter indicates no statistical difference.
|
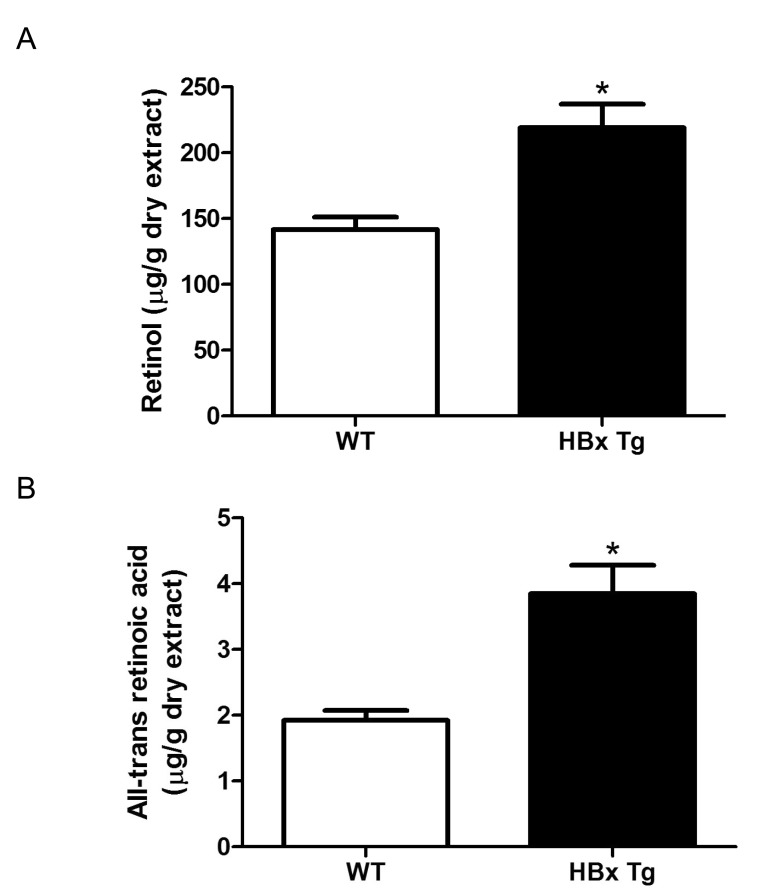
We also measured hepatic retinol and ATRA levels in HBx Tg mice. Both retinol and ATRA levels in liver were higher in HBx Tg mice than in WT control mice (Fig. 3).
The effect of metformin treatment on hepatic CRBP-I and on the development of hepatocellular carcinoma (HCC) in HBx Tg mice
In order to test whether chronic metformin treatment of HBx Tg mice affects the incidence of HCC, metformin was administered to the mice from 6 weeks of age till 18 months. Representative histological images of HCC in HBx Tg mice are presented in Fig. 4. Although metformin treatment of HBx Tg mice significantly decreased the fasting blood glucose (FBS) and serum insulin levels compared with un-treated HBx Tg mice, the incidence of HCC was not affected by metformin treatment (Table 1). Hepatic CRBP-I protein level was increased by metformin treatment in HBx Tg mice compared to that in un-treated HBx Tg mice, whereas Akt level was decreased by the treatment (Fig. 5).
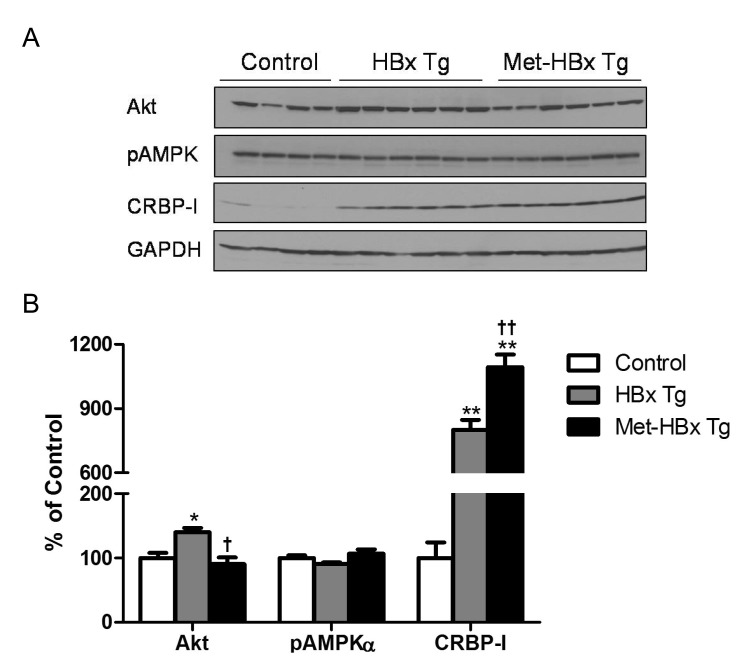 | Fig. 4Hepatic expression levels of Akt, CRBP-I, and pAMPK in WT and HBx Tg mice, and metformin-treated HBx Tg mice (Met-HBx Tg). The representative immunoblotting images (A) and the levels of protein expression were presented (% of control) (B). *p<0.05; **p<0.01 vs. WT mice. †p<0.05; ††p<0.01 vs. HBx Tg mice.
|
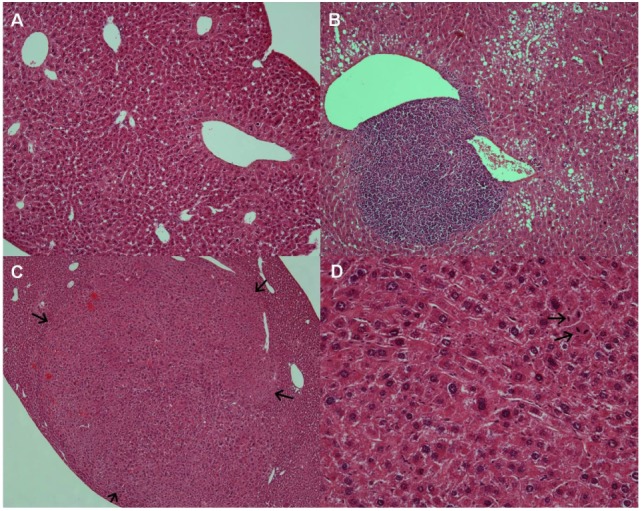 | Fig. 5The fatty liver change and the development of HCC in HBx Tg mice. Hematoxylin and eosin-stained representative images are presented. (A) Relatively normal-looking portion in the liver of HBx Tg mice (×100). (B) Fatty changes and chronic inflammatory cell infiltrations are noted in the dysplastic liver of HBx Tg mice (×100). (C) Microscopic appearance of grossly identified HCC. The un-encapsulated and well differentiated HCC lesion has a trabecular pattern and is compressing the surrounding dysplastic hepatocytes (×40). Arrows indicate the boundary of HCC. (D) Microscopic appearance of grossly identified HCC shows nuclear pleomorphism, nucleomegaly and frequent mitoses (×200). Arrows indicate mitosis.
|

Go to :
DISCUSSION
In the present study, we evaluated hepatic CRBP-I and retinoid pathway in HBx Tg mice which have characteristic metabolic phenotypes such as fatty liver, IGT, and IR and are also prone to HCC [2,3,5]. Hepatic CRBP-I expression was markedly increased in HBx Tg mice, but not in db/db mice and HF-fed WT mice, compared to WT control mice. HBx Tg mice showed also an up-regulation of genes for RA synthesis and of some genes for RA signaling (RXRα, Tgm2, and a strong tendency to an increase in FABP5 expression). In addition, hepatic retinol and ATRA levels were higher in HBx Tg mice than in WT control mice. We also evaluated the effect of metformin treatment on the development of HCC in HBx Tg mice. Metformin treatment of HBx Tg mice further increased CRBP-I protein level in the liver and did not affect the incidence of HCC.
A study of HCC patients has shown that CRABP and fish type CRBP were increased in HCC tissue compared to surrounding tissue, with similar expression of CRBP [23], although isotypes of retinoid-binding proteins were not described in detail. However, other studies revealed that the expression of CRABP-II (but not of CRABP-I) would enhance the transcriptional activity of RAR [11,24], leading to the inhibition of tumor cell proliferation by RA [25]. Considering contradictory data between studies as discussed above, previous study findings may not be generalized for clinical applications. In addition, data about CRBP-I and CRABP-II in HCC models are lacking. To the best of our knowledge, this is the first study to show the marked increase in hepatic CRBP-1 expression in HBx Tg mice.
Studies have reported a decreased retinoid level in hepatoma tissue and/or cancer-prone hepatic tissue [8,26]. In addition, the anticarcinogenic activity of retinoids in many animal models has been documented for various types of cancers [27,28]. In patients with HCC, an acyclic retinoid, polyprenoic acid, prevented second primary hepatomas after successful initial treatment [29]. In a normal liver, hepatic stellate cells (HSCs) store up to 80% of the total body vitamin A in specific lipid droplets [30]. In the presence of chronic active liver diseases, HSCs shows a different phenotype characterized by the progressive loss of lipid droplets, with an excess of proliferation [30]. However, in mice lacking LRAT, despite the hepatic retinyl ester lipid droplets are nearly absent, diethylnitrosamine-induced HCC mass was shown to be decreased. Other groups have reported a trend towards higher hepatic levels of RA in LRAT-deficient mice [31,32]. Furthermore, McCormick et al. [33] reported that ATRA and two other synthetic retinoids could promote liver carcinogenesis in carcinogentreated mice when administered at doses that inhibit cancer induction in other tissues.
Thus, the association of HCC with tissue levels of CRBPs and retinoids in liver seems to be more complex than expected. Further studies using various disease models are required.
HBx Tg mice seem to be a good model for studying HCC, fatty liver, and IR [2,3,5]. The increased risk of HCC in patients with diabetes or NAFLD has been reported [6,34]. In addition, many studies showed that retinoids and CRBP-I are involved in the regulation of glucose and lipid homeostasis [12,15]. However, less is known about hepatic CRBP-I and retinoid levels in HBV-related models. Our findings indicated that hepatic CRBP-I was markedly increased in HBx Tg mice, independent of fatty liver or IR. We also observed an increase in hepatic retinol and ATRA level in HBx Tg mice. Given these contradictory results of the present study to the previous studies [8,32,35], we thought that a better knowledge of the CRBP-I and retinoid metabolism was needed for various models of chronic liver disease and HCC.
While holo-CRBP-I presents its ligand directly to LRAT, apo-CRBP-I inhibits LRAT reaction and stimulates the hydrolysis of retinyl esters by its hydrolase [11]. Thus, the physiologically relevant scenario suggests that, in the HBx mice, apo-CRBP-I will both inhibit retinol esterification and stimulate liberation of retinol from stored retinyl esters, thereby allowing retinol to be available for the synthesis of RA. Although RA itself functions as a master determinant of its synthesis and catabolism by inducing CRBP, by enhancing retinol esterification via inducing LRAT and by inducing catabolic enzyme [36], our study findings suggest that these regulating pathways seem to be disorganized in HBx Tg mice.
In agreement with previous studies [36], our findings showed that increases in RA biosynthesis from retinol accompanied an increase in CRABP-II expression. RA can act as PPARδ ligand, resulting in stimulation of cell growth and inhibition of apoptosis in certain cell types [22]. As intracellular lipid-binding proteins, CRABP-II specifically cooperates with RARs, and FABP5 functions in conjunction with PPARδ [22,37]. Tgm2 plays a diverse role in a variety of cellular processes, including cell death, proliferation, differentiation, and migration. Various factors including RA, transcription factors, cytokines, and tumor growth factor β1 can induce Tgm2 [38]. Further studies are required to clarify whether the up-regulations of some of RA target genes (Tgm2, FABP5) contribute to the development of HCC.
Many studies have shown an inverse association between HCC risk and metformin therapy among patients with type 2 diabetes [16,17]. Because CRBP-I up-regulation and HCC development may also be related with metabolic changes in the liver, such as IR and steatosis, of HB Tg mice [2], we assessed the effect of chronic metformin treatment. However, metformin treatment had no effect on HCC development in HBx Tg mice in the present study and did not suppressed hepatic CRBP-I. Because HBV infection, rather than metabolic diseases, is a stronger risk factor for HCC, the chemopreventive effect of metformin might be attenuated in this mice model.
The one of major limitations of the present study is that we did not clarify the effect of metformin on retinoid pathway. Because metformin did not have HCC-suppressive effect in our experimental model, we did not performed further detailed experiments about the relationship between metformin and retinoid pathways in the HCC-prone HBx Tg mice.
In conclusion, hepatic CRBP-I level was markedly up-regulated in HCC-prone HBx Tg mice with some characteristic changes in retinoid metabolism and signaling pathways. Neither hepatic CRBP-I level nor the development of HCC was suppressed by metformin treatment of these transgenic mice. Our findings suggested that an over-simplified view on retinoid physiology in chronic liver disease should be avoided and further studies using various disease models are required for better knowledge of retinoids.
Go to :
 XML Download
XML Download