

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Insulin-dependent Diabetes mellitus (IDDM) is an autoimmune disease that selectively destroys insulin-producing beta cells in the pancreatic islets of Langerhans. Strong experimental evidence suggests that nitric oxide (NO) is produced by proinflammatory cytokines, such as tumor necrosis factor (TNF)-α, interferon (IFN)-γ, and interleukin (IL)-1β and plays an important role in the development of pancreatic beta cell destruction by stimulating production of oxygen radicals [1-3]. Further, IFN-γ, TNF-α and IL-1β synergistically activate inducible NO synthase (iNOS) mRNA expression and NO production [1,4]. Studies on transgenic mice have shown that NO is important in beta cell destruction [5,6]. Specifically, transgenic mice expressing mouse iNOS cDNA under the control of the insulin promoter have been shown to develop diabetes, whereas treatment with aminoguanidine, an iNOS inhibitor, was shown to prevent or delay the development of diabetes [5]. Further, it was shown that IL-1β fails to induce iNOS mRNA expression or increase nitrite formation in islets isolated from iNOS knockout mice [6]. iNOS knockout mice further display reduced sensitivity to multiple low-dose streptozotocin-induced diabetes [6]. Therefore, inhibition of high-output NO production by blocking iNOS production or activity may be a useful strategy for the treatment of inflammatory disorders, including IDDM.
Radicicol is a macrocyclic fungal antibiotic originally isolated from the fungus Monosporium bonorden and inhibits angiogenesis [7,8]. Radicicol induces reversal of the transformed phenotype of src-transformed cells and exhibits p60v-src tyrosine kinase inhibitor activity [9,10]. Radicicol also blocks the activation of the mitogen-activated protein kinase/extracellular signal-regulated kinase (MAPK/ERK) pathway through destabilization of Raf kinase [11]. It has been reported that radicicol suppresses the expression of mitogen-inducible cyclooxygenase, which is important in inflammatory responses [12]. We have shown that expression of iNOS, another important mediator of inflammatory responses, p38 kinase pathway activation, and NF-κB activation were inhibited by radicicol in LPS-stimulated macrophages [13]. In the present study, we investigated the effects of radicicol on the regulation of iNOS, NF-κB activity, p65 nuclear translocation, and p44/42 activity in proinflammatory cytokine-stimulated MIN6N8a cells, a mouse pancreatic beta cell line.
METHODS
Cell culture and Reagents
MIN6N8a cells, SV40 T-transformed insulinoma cells derived from NOD mice, were grown in Dulbecco's Modifed Eagle's Medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 100 U/ml of penicillin, 100 µg/ml of streptomycin, and 50 µM 2-mercaptoethanol. For each experiment, cells (5×105 cells/ml) were plated in 100-mm dishes. Radicicol, SN50, PD98059 (2'-amino-3'-methoxyflavone), and SB203580 (4-(4-fluorophenyl)-2-(4-methylsulfinylphenyl)-5-(4-pyridyl) 1H-imidazole) were purchased from CalBiochem (San Diego, CA). Anti-iNOS and anti-p65 antibodies were purchased from Upstate Biotechnology (Lake Placid, NY) and Santa Cruz Biotechnology Inc (Santa Cruz, CA), respectively. Antibodies against phospho-p44/42 and p44/42 were purchased from Cell Signaling Technology, Inc. (Beverly, MA).
Nitrite determination
MIN6N8a cells were treated with the indicated concentrations of radicicol in the presence of cytokine mixture (CM: TNF-α, 500 U/ml; IFN-γ, 100 U/ml; IL-1β, 10 U/ml) for 48 h. Culture supernatants were collected, and the accumulation of NO2- in culture supernatants was measured as an indicator of NO production in the medium as previously described [14,15].
Immunofluorescence statining
MIN6N8a cells were treated with radicicol (100 ng/ml) in the presence of CM on a cover slide in 12-well plates. Cells were rinsed 3 times with PBS, fixed with 4% paraformaldehyde for 10 min at room temperature, and rinsed again. Cells were then blocked with 1% bovine serum albumin, followed by the addition of the primary antibody. After extensive washing with Tris-buffered saline, fluorescein isothiocyanate-conjugated IgG was added. Following incubation, the slides were rinsed, mounted, and viewed at 488 nm on a confocal microscope (FV300, Olympus, Japan).
RT-PCR
Total RNA was isolated with TRI Reagent (Molecular Research Center, Cincinnati, OH, USA). Forward and reverse primer sequences were as follows: iNOS: 5'-CTG CAG CAC TTG GAT CAG GAA CCT G-3', 5'-GGG AGT AGC CTG TGT GCA CCT GGA A-3'; and β-actin: 5'-TGG AAT CCT GTG GCA TCC ATG AAA C-3', 5'-TAA AAC GCA GCT CAG TAA CAG TCC G-3'. Equal amounts of RNA were reverse-transcribed into cDNA with oligo(dT)15 primers. PCR was performed with cDNA and each primer. Samples were heated to 94℃ for 5 min and cycled 30 times at 94℃ for 1 min, 55℃ for 1.5 min, and 94℃ for 1 min, after which an additional extension step at 72℃ for 5 min was conducted. PCR products were separated by 8% SDS-PAGE, followed by staining in ethidium bromide. The iNOS and β-actin primers produced amplified products of 311 and 349 bp, respectively.
Western immunoblot analysis
Whole cell lysates were separated by 10% SDS-PAGE and then electrotransferred to nitrocellulose membranes (Amersham International, Buckinghamshire, UK). The membranes were then preincubated for 1 h at room temperature in Tris-buffered saline (TBS), pH 7.6 containing 0.05% Tween-20 and 3% bovine serum albumin, followed by incubation with iNOS, phosphorylated ERK1/2, and phospho-ERK1/2 (Thr202/Tyr204)-specific antibodies. Immunoreactive bands were detected by incubation with conjugates of anti-rabbit IgG with horseradish peroxidase and enhanced chemiluminescence reagent (Amersham).
Electrophoretic mobility shift assay (EMSA)
EMSA was performed as described in previous literature [16,17]. Nuclear extracts were prepared as previously described [18]. The double-stranded oligonucleotides were end-labeled with [γ-32P]-ATP. Nuclear extracts (5 µg) were incubated with poly (dI-dC) and the [32P]-labeled DNA probe in binding buffer (100 mM KCl, 30 mM HEPES, 1.5 mM MgCl2, 0.3 mM EDTA, 10% glycerol, 1 mM DTT, 1 mM PMSF, 1 µg/ml of aprotinin, and 1 µg/ml of leupeptin) for 10 min. DNA-binding activity was separated from free probe by 4% SDS-PAGE in 0.5× TBE buffer. Following electrophoresis, the gel was dried and subjected to autoradiography.
Transient transfection of MIN6N8aMIN6N8a cells
Vector constructions were performed as previously described [19]. MIN6N8aMIN6N8a cells were transfected using the DEAE-dextran method, diluted to 5×105 cells per 1 ml of complete media, plated on 24-well plates, and then incubated in the presence of 5% CO2 at 37℃ for 24 h. The transfectants were then treated with CM and radicicol for 18 h, after which cells were lysed with lysis buffer. The lysates were centrifuged (12,000×g for 10 min at 4℃), and the supernatant was assayed for CAT enzyme expression using a CAT ELISA kit (Roche Molecular Biochemicals, Mannheim, Germany) according to the manufacturer's instructions.
Statistical analysis
The mean±SD was determined for each treatment group in a given experiment. When significant differences occurred, treatment groups were compared to the vehicle controls using a Dunnett's two-tailed t test [20].
RESULTS
Effect of radicicol on iNOS expression in cytokine-stimulated MIN6N8a cells
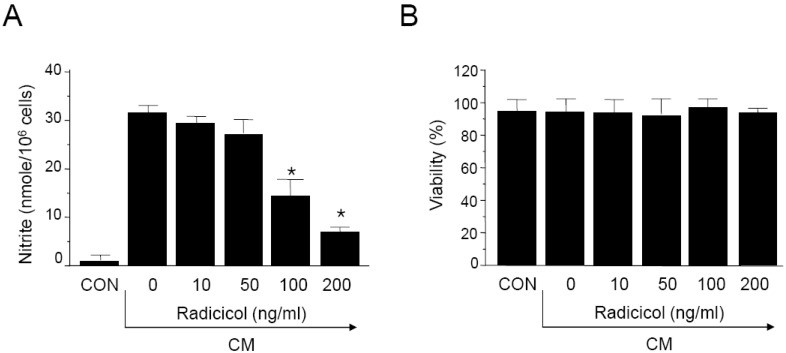
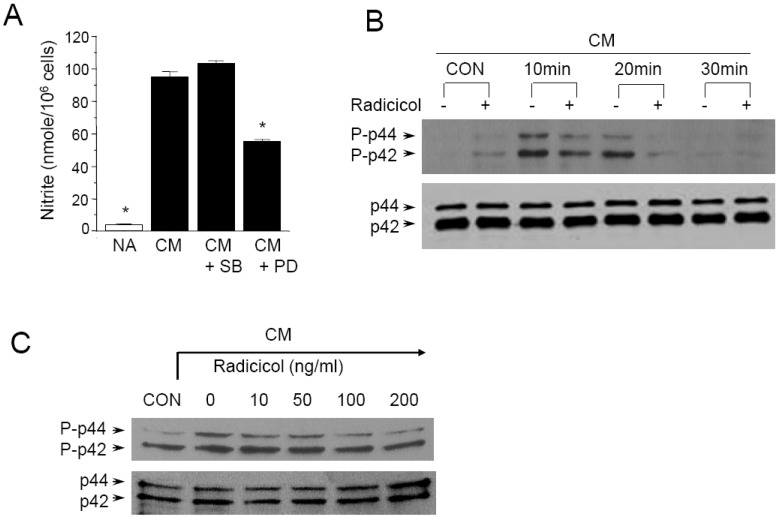
Cytokines, including IL-1β, IFN-γ, and TNF-α are known to induce or potentiate iNOS expression and NO production [1-3]. We investigated the effects of radicicol on NO production in cytokine-stimulated MIN6N8a, a mouse pancreatic beta cell line. Cytokine mixture (CM: TNF-α, 500 U/ml; IFN-γ, 100 U/ml; IL-1β, 10 U/ml) increased production of nitrite ≥15-fold over basal levels in MIN6N8a cells (Fig. 1A). This induction of nitrite generation by CM was inhibited by radicicol in a dose-dependent manner. No effect on cell viability was observed in any of the treatment groups, and it always exceeded 90% as determined by trypan blue staining (Fig. 1B).
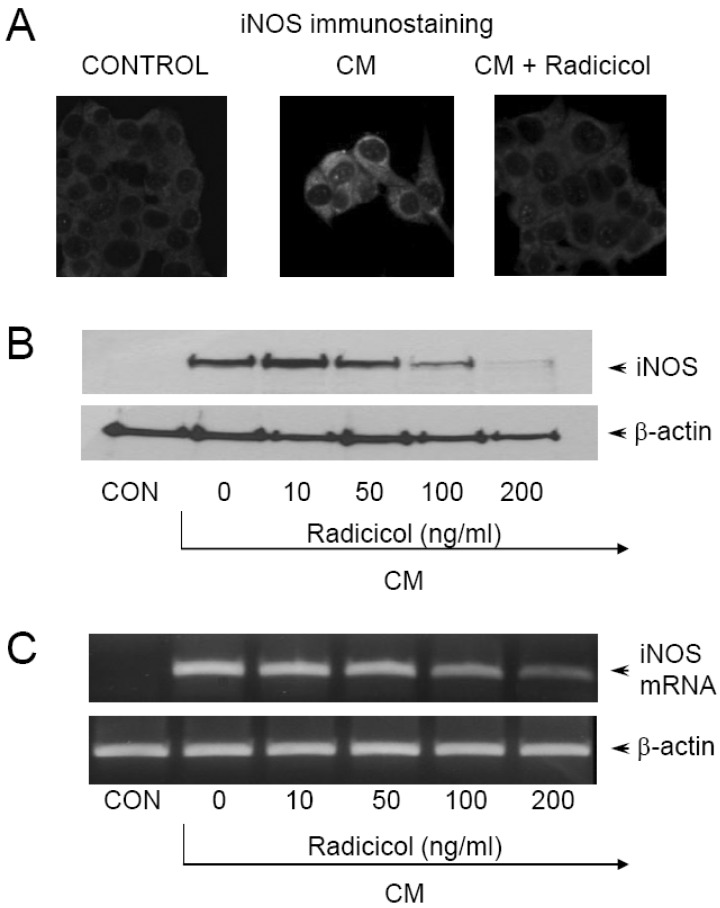
To investigate the effects of radicicol on iNOS production, expression of iNOS was measured by immunohistochemical staining. MIN6N8a cells (5×105 cells/ml) were incubated with radicicol (100 ng/ml) in the presence of CM for 24 h on a cover slide in 12-well plates. Cells were then subjected to immunohistochemical staining using an antibody specific for murine iNOS. Immunofluorescence staining of iNOS showed that radicicol inhibited iNOS production (Fig. 2A). After MIN6N8a cells were exposed to radicicol in the presence of CM, the expression level of iNOS was monitored by Western immunoblot analysis. As shown in Fig. 2B, iNOS protein production was inhibited by radicicol treatment. Consistent with these findings, radicicol inhibited the production of iNOS mRNA in CM-stimulated MIN6N8a cells (Fig. 2C). Control β-actin was constitutively expressed and was not affected by radicicol treatment. These results indicate that radicicol reduces gene expression of iNOS, which is involved in pancreatic beta cell destruction.
Inhibition of NF-κB/Rel activation by radicicol in CM-stimulated MIN6N8a cells
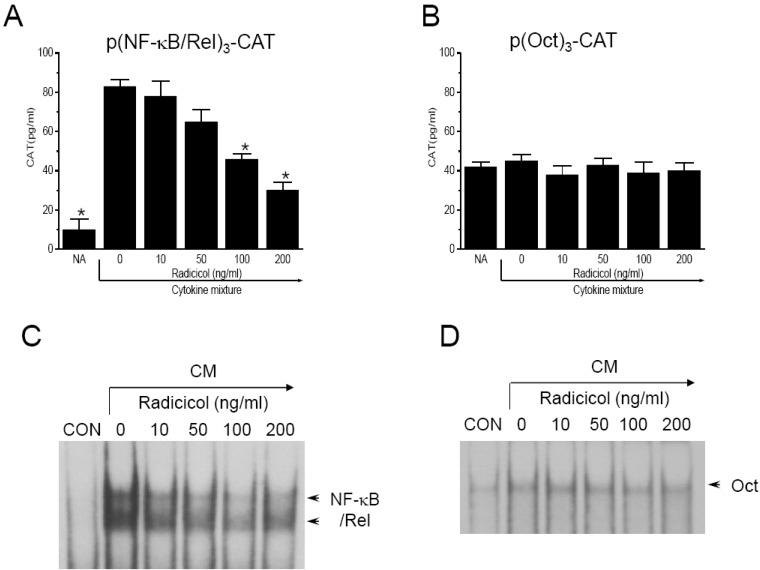
Since it has been reported that protein binding at NF-κB/Rel-binding sequences is necessary for induction of iNOS expression [21], we assessed the effect of radicicol on NF-κB/Rel activation using a transient transfection assay. MIN-6N8a cells were transiently transfected with p(NF-κB/Rel)3-CAT showed reduced gene expression of CAT in response to radicicol treatment in the presence of CM (Fig. 3A). Although CM treatment significantly increased CAT expression by MIN6N8a cells, CM-induced CAT expression was inhibited by radicicol treatment. Oct, a transcription factor that binds to the promoter of the iNOS gene, moderated CAT activity and was not influenced by either CM or radicicol treatment (Fig. 3B). Transcriptional activation of the NF-κB/Rel transcription factor is preceded by its binding to DNA. Therefore, we further analyzed the effect of radicicol on activation of NF-κB/Rel, whose binding motif is in the promoter of the iNOS gene, using EMSA. MIN6N8a cells treated with CM displayed a marked increase in NF-κB/Rel binding to its cognate site. Further, induction of NF-κB/Rel binding was inhibited by radicicol (Fig. 3C). Radicicol had no effect on Oct DNA-binding activity (Fig. 3D). The specificity of the retarded bands was confirmed by the addition of excess 32P-unlabeled double-stranded κB or Oct (data not shown). These results indicate that radicicol reduces the DNA-binding activity and transcriptional activation of NF-κB/Rel, which is important in the regulation of iNOS gene expression.
Inhibition of p65 nuclear translocation by radicicol in CM-stimulated MIN6N8a cells
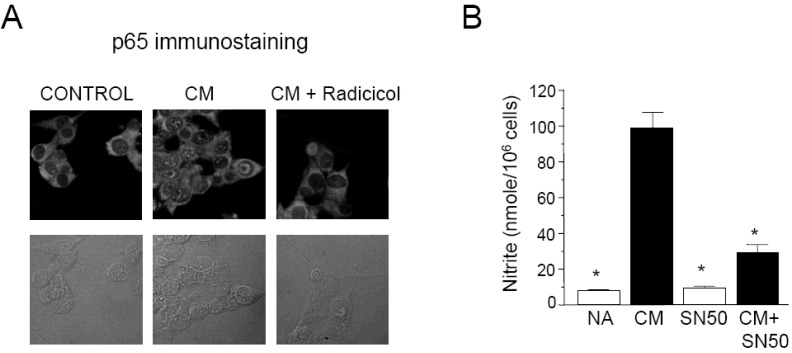
To further investigate whether or not radicicol inhibits nuclear translocation of p65, a component of NF-κB/Rel, we performed immunofluorescence staining. CM-stimulated MIN6N8a cells showed marked p65 staining in nuclei, whereas unstimulated cells showed weaker nuclear p65 expression along with stronger staining in the cytoplasm. Radicicol treatment significantly inhibited CM-induced nuclear translocation of p65 (Fig. 4A). These results indicate that radicicol reduces the nuclear translocation and DNA-binding activity of NF-κB/Rel, which is important in the regulation of iNOS gene expression.
We further confirmed the involvement of p65 nuclear translocation and NO production using SN50, a NF-κB/Rel nuclear translocation inhibitor. SN50, a cell-permeable inhibitor peptide, was reported to inhibit nuclear translocation of NF-κB/Rel [22]. Treatment of MIN6N8a cells with SN50 blocked CM-induced nitrite generation (Fig. 4B). Collectively, these results demonstrate that NF-κB/Rel plays an important role in the activation of iNOS gene expression by CM in pancreatic beta cells, and it is inhibited by radicicol.
Inhibition of p44/42 phosphorylation by Radicicol in CM-stimulated MIN6N8a cells
Since p44/42 and p38 kinases are possible targets of radicicol, we further determined whether or not these kinase pathways are involved in CM-induced NO production. The p44/42 and p38 kinase pathways were specifically blocked when MIN6N8a cells were challenged with CM. PD98059 is a specific inhibitor of mitogen activated protein kinase/extracellular signal-regulated kinase 1 (MEK-1), which is responsible for ERK1/2 activation. SB203580, a bicyclic imidazole compound, is a specific inhibitor of p38. PD98059 inhibited CM-induced production of nitrite, whereas SB20-3580P had no inhibitory effect (Fig. 5A). These results suggest that the p44/42 kinase pathway is important in the regulation of NO production in MIN6N8a cells by CM.
We further assessed the effect of radicicol on p44/42 phosphorylation in CM-stimulated MIN6N8a cells. CM-induced phosphorylation of p44/42 peaked at 10 min, was maintained until 20 min, and then decreased at 30 min (Fig. 5B). On the other hand, radicicol strongly inhibited p44/42 phosphorylation. A dose-response experiment further showed that radicicol inhibited CM-induced p44/42 phosphorylation in a dose-dependent manner (Fig. 5C). Collectively, this series of experiments indicates that p44/42 kinases are important in the regulation of iNOS in CM-stimulated MIN6N8a cells, and they are inhibited by radicicol.
DISCUSSION
In a previous study, we showed that production of NO, an important mediator of inflammatory responses, is inhibited by radicicol in LPS-stimulated macrophages [13]. Since proinflammatory cytokines are known to induce the expression of iNOS mRNA and production of NO, resulting in cell death of beta cells [1-3], we investigated whether or not radicicol suppresses iNOS gene expression induced by cytokines. In the present study, we found that radicicol inhibits iNOS gene expression in cytokine-stimulated MIN6N8a beta cells.
NF-κB/Rel plays an important role in cytokine-induced beta cell destruction, as cell death is blocked in beta cells by inhibition of NF-κB/Rel activation [23,24]. Conditional and specific blockage of NF-κB/Rel protects pancreatic beta cells against diabetes induced by multiple low doses of streptozotocin [25]. We observed that NF-κB/Rel was positively regulated by CM for induction of iNOS gene expression, whereas radicicol significantly inhibited the CM-induced NF-κB/Rel transcriptional activation and DNA-binding activity (Fig. 3). NF-κB/Rel is a regulator of many genes involved in inflammatory responses, including iNOS [21]. NF-κB/Rel exists in the cytoplasm of unstimulated cells in an inactive form bound to IκB, an inhibitor of NF-κB/Rel. Activation by external stimuli including lipopolysaccharide and cytokines results in the phosphorylation of IκB, which releases the active DNA-binding form of NF-κB/Rel for translocation to the nucleus to bind κB motifs in the promoter regions of various genes. EMSA studies showed strong induction of two separate κB-binding complexes by CM. Further, radicicol inhibited activation of both of the binding complexes (Fig. 3C). It has been shown that p50 proteins have DNA-binding activity, whereas p65 proteins contain transactivation domains in their C termini and are thus able to activate the transcription of target genes [26]. Immunofluorescence staining of p65, the active component of NF-κB/Rel, confirmed nuclear translocation of p65 induced by CM, whereas radicicol inhibited nuclear translocation of p65 (Fig. 4A).
We also showed that radicicol significantly inhibits the ERK1/2 pathway in MIN6N8a cells. It has been reported that radicicol blocks the activation of the MAPK/ERK pathway through destabilization of Raf kinase [11,27]. ERK activity is required for iNOS gene expression in insulin-producing INS-1E cells [28]. In the present study, we investigated the involvement of the ERK1/2 and p38 pathways in iNOS regulation in a mouse insulinoma cell line stimulated with cytokines. Two inhibitors, PD98059 and SB203580, which selectively inhibit the ERK1/2 and p38 pathways, respectively, were used to address this issue. Data presented herein clearly show that the ERK1/2 pathway is involved in iNOS regulation in MIN6N8a cells. Specifically, PD98059 inhibited NO production in response to CM. ERK has been reported to be involved in NF-κB/Rel-mediated transcription of iNOS, indicating that ERK regulates iNOS gene expression by increasing the transactivation capacity of NF-κB/Rel [28].
In summary, these experiments demonstrate that radicicol inhibits CM-induced iNOS gene expression in MIN6N8a cells. Based on our findings, the most likely mechanism that can account for this biological effect involves inhibition of NF-κB and the ERK1/2 kinase pathway. Due to the critical role that NO release plays in mediating inflammatory responses in pancreatic beta cells, the inhibitory effects of radicicol on iNOS expression suggest that this anti-fungal may represent a useful anti-diabetic agent.
 XML Download
XML Download