

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Curcumin (diferuloylmethane), a polyphenol derived from the plant turmeric (Curcuma longa Linn), is known for its antioxidant and anti-inflammatory properties [1]. Moreover, quite a few studies, including our own, have shown that curcumin exhibits anticancer activities against various tumors, including pancreatic, colon, ovarian, and breast cancers, by modulating multiple signaling pathways [2,3]. Curcumin has been reported to increase p53 signaling, which upregulates the transcription of downstream genes such as p21 and Bax to induce apoptosis in a variety of cancer cells [2]. In addition, curcumin causes cancer cell apoptosis, in part via suppression of Notch1 signaling [4-8]. However, in this case, little has been reported about the underlying molecular mechanisms by which Notch1 expression is modulated upon curcumin treatment.
Notch receptors (Notch1~4) are large single-pass type I transmembrane proteins that regulate fate determination, differentiation, proliferation, motility, and survival of various cell types in different tissues [9]. Many studies have shown the important role of Notch signaling not only in normal embryonic development and postnatal tissue homeostasis, but also in cancer [9]. Notch1 has been shown to act as either a tumor suppressor or an oncogene in human cancer [10]. When acting as an oncogene, the Notch1 receptor, ligands, and target genes are significantly upregulated, resulting in stimulation of cellular proliferation, prevention of differentiation, and inhibition of apoptosis [10,11]. Recent evidence shows the role of Notch1 as a bio marker in triple-negative breast cancer [12]. Moreover, the abrogation of aberrant Notch1 signaling was demonstrated to lead to apoptosis of breast cancer cells [13]. Therefore, targeting Notch1 provides a potential novel strategy for therapeutic intervention in breast cancer.
TP53 is a key tumor suppressor gene encoding the p53 transcription factor that is activated in response to a variety of cellular insults such as DNA damage, hypoxia, and oncogene activation [14-16]. Activated wild-type p53 regulates the transcription of target genes after binding to a p53 DNA-binding consensus sequence and exerting anti-proliferative functions by inducing DNA repair, cell growth arrest, and apoptosis in damaged cells [17-19]. TP53 is mutated in >50% of human cancers, and mutant p53 proteins not only lose tumor-suppressive roles of wild-type p53 and possess dominant-negative activities, but also acquire new oncogenic gain-of-function activities [20]. So far, several genes have been reported to be associated with the gain-of-function properties of mutant p53, contributing to the development and progression of tumorigenesis [21,22]. For example, ID2, ATF3, MST1, and Bcl-xL are downregulated by mutant p53, whereas MDR-1, c-Myc, TERT, VEGFA, and NF-κB are upregulated by mutant p53. Although many targets of mutant p53 have been discovered, more are believed to exist within mutant p53 signaling pathway. Recently, wild-type p53 was demonstrated to affect Notch1 expression [23-26]. However, little has been reported regarding the effect of mutant p53 on Notch1 expression in breast cancer cells.
Interestingly, we have found one p53-binding site on human Notch1 gene promoter. Therefore, the purpose of this study was to investigate whether Notch1 is a downstream target gene of mutant p53 in human breast cancer cell line MDA-MB-231, which harbors a p53 missense mutation (expressing p53-R280K), (35, 40), and to characterize the role of curcumin as an inhibitor of mutant p53-Notch1 axis in MDA-MB-231 cells.
METHODS
Materials
Curcumin and mouse IgG were purchased from Sigma (St Louis, MO). Mouse monoclonal anti-p53 and anti-β-actin antibodies were obtained from Calbiochem (San Diego, CA) and Abcam (Cambridge, MA), respectively. Rabbit polyclonal anti-cleaved Caspase-3 and anti-cleaved Notch1 (c-Notch1) antibodies were purchased from Cell signaling Technology (Beverly, MA) and Milipore (Billerica, MA), respectively. Rabbit polyclonal anti-total Notch1 (t-Notch1), anti-PARP, anti-Bcl-2 and mouse monoclonal anti-Caspase-3 and c-Myc antibodies were acquired from Santa Cruz Biotechnology (Santa Cruz, CA). Horseradish peroxidase-conjugated goat anti-rabbit IgG and goat anti-mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Rabbit anti-Hes1antibody (Chemicon, AB5702) was obtained from Chemicon (Temecula, CA).
Cell culture
MDA-MB-231 cells were obtained from the American Type Culture Collection (Manassas, VA) and cultured in Dulbecco's modified eagle's medium (DMEM: GIBCO BRL, Grand Island, NY) containing 10% heat-inactivated fetal bovine serum (FBS: GIBCO BRL, Grand Island, NY) and 1% antibiotics. Cells were maintained in a humidified 5% CO2 atmosphere at 37℃.
Reverse transcription polymerase chain reaction (RT-PCR)
Total RNAs were isolated from MDA-MB-231 cells with TRIzol reagent kit (Invitrogen, Carlsbad, CA). cDNA synthesis was performed using 2 µg of total RNA with a reverse transcription kit (Promega, Madison, WI). The oligonucleotide primers for PCR were designed as follows: β-actin, 5'-GACTACCTCATGAAGATC-3' and 5'-GATCCACATCTGCTGG AA-3'; p53, 5'-GGCCCACTTCACCGTACTAA-3' and 5'-GTGGTTTCAAGGCCAGATGT-3'; Notch1, 5'-GCAACAGCTCCTTCCACTTC-3' and 5'-GCCTCAGACACTTTGAAGCC-3'; Notch2, 5'-CCCAATGGGCAAGAAGTCTA-3' and 5'-CACAATGTGGTGGTGGGATA-3'; Notch3, 5'-TCTTGCTGCTGGTCA TTCTC-3' and 5'-TGCCTCATCCTCTTCAGTTG-3'; Notch4, 5'-TATTCTCATTGCCGGAGCCTCTCGGGAGTA-3' and 5'-ACCCTCTCCTCCTTGGTTTATGGGCATTTC-3'; Hes1, 5'-AGCACAGAAAGTCATCAAAGCC-3' and 5'-TTCATGCACTCGCTGA AGCC-3'.
Western blot analysis
Harvested cells were lysed in a lysis buffer (40 mM Tris-Cl, 10 mM EDTA, 120 mM NaCl and 0.1% NP-40 with protease inhibitor cocktail (Sigma, St Louis, MO). A constant protein concentration (30 µg/lane) was used. The proteins were separated by SDS/PAGE and transferred to a nitrocellulose membrane (Amersham Pharmacia Biotech, Piscataway, NJ). The membrane was blocked with 5% skim milk in phosphate buffered saline (PBS) containing 0.1% Tween-20 for 1 h at room temperature and probed with appropriate antibodies. The signal was developed using the enhanced chemiluminescence (ECL) detection system (Amersham Pharmacia Biotech, Piscataway, NJ). β-actin served as the loading control. Band intensities were quantified using MetaMorph software (Molecular Devices, Union City, CA).
Construction of Notch1 promoter-driven reporter vectors and pCMV-p53 expression vectors
The human Notch1 promoter regions -3288 to -3, 2368 to -3, -1275 to -3, and -486 to -3 (numbered relative to the ATG) were amplified by PCR from genomic DNA isolated from human microvascular endothelial cells with the following primer pairs: 5'-GACGCGTGCAAGAGCCAAGATGAACTCCTGC-3' and 5'-CAGATCTCTCCCCACCGGCTGCCCTCTG-3' (3286 bp); 5'-GACGCGTGACAAAGAGGAATGGGGAGC-3' and 5'-CAGATCTCTCCCCACCGGCTGCCCTCTG-3' (2366 bp); 5'-GACGCGTTGCCATCGCACTCACCACCCTGTG-3' and 5'-CAGATCTCTCCCCACCGGCTGCCCTCTG-3' (1273 bp); 5'-GACGCGTAACGAGAAGTAGTCCCAGGC-3' and CAGATCTCTCCCCACCGGCTGCCCTCTG-3' (484 bp). The PCR amplified products were subcloned into the pGEM-T easy vector (Promega, Madison, WI) and checked by automatic DNA sequencing (Cosmo, Seoul, Korea). The cloned sequences were released by restriction digestion with Mlu I and Bgl II and subcloned into the pGL3 basic vector (Promega, Madison, WI) at the same sites. pCMV-p53 expression vector, which harbors wild-type p53, was purchased from Clontech Laboratories, Inc. Site-directed mutagenesis was performed to construct pCMV-mutant p53, which contains mutant p53-R280K, from pCMV-p53 by the QuickChange mutagenesis kit (Stratagene, La. Jolla, CA) according to the manufacturer's instructions. The oligonucleotide primers used in site-directed mutagenesis were designed as follows: p53-R280K, 5'-TGTGCCTGTCCTGGGAAAGACCGGCGCACAGAG-3' and 5'-CTCTGTGCGCCGGTCTTTCCCAGGACAGGCACA-3'. The sequences of the mutation construct were checked by automatic DNA sequencing (Cosmo, Seoul, Korea).
Transient transfection and reporter gene analysis
For the luciferase assays, luciferase reporter plasmids with pCMV-β-galactosidase were transfected into MDAMB-231 cells by using X-tremeGene HP DNA transfection reagent (Roche Applied Science, Indianapolis, IN). Cell lysates were then subjected to β-galactosidase enzyme assay and were tested for luciferase activity by using an assay kit (Promega, Madison, WI) and luminometer (Turner Designs, Sunnyvale, CA).
Transient transfection of siRNA
We designed and synthesized double-stranded siRNA oligonucleotides (Bioneer, Daejeon, Korea) against p53 (5'-GACUCCAGUGGUAAUCUACTT-3' and 5'-GUAGAUUACCACUGGAGUCTT-3') and Notch1 (5'-GAACGGGGCUAACAAAGAUTT-3' and 5'-AUCUUUGUUAGCCCCGUUCTT-3'). A non-silencing fluorescein labeled siRNA (Bioneer, Daejeon, Korea) was used as control for transfection efficiency as well as for monitoring the effect of silencing during all experiments. Transfection of MDA-MB-231 cells with siRNA (200 nM) was performed using Oligofectamine (Invitrogen, Carlsbad, CA) according to the recommendations of the manufacturer.
Nuclear protein extraction and electrophoretic mobility shift assay (EMSA)
Nuclear extracts from MDA-MB-231 cells were prepared and analyzed by EMSA as previously described [3]. Binding reactions containing equal amounts of nuclear extracts (10 µg) and biotin-labeled oligonucleotide probes were performed for 20 min in binding buffer (10 mM Tris-HCl, 50 mM KCl, 1 mM DTT, 5 mM MgCl2 and 0.05% NP-40) at room temperature. The binding reactions were analyzed using 6% native PAGE. After blotting onto a nylon membrane, labeled oligonucleotides were detected with the Light Shift Chemiluminescent EMSA Kit following the instructions of the manufacturer (Pierce, Rockford, IL). The oligonucleotide sequences for EMSA were as follows: p53-binding site on human Notch1 promoter (Bioneer, Daejeon, Korea), 5'-AGCCTCACTAGTGCCTCGGCCGCGGGAGGGAGCGC AAGGGCGCGG-3' and 5'-CCGCGCCCTTGCGCTCCCTCCCGCGGCCGAGGCACTAGT GAGGCT-3'.
Chromatin immunoprecipitation assay (ChIP)
Chromatin immunoprecipitation analysis was performed on MDA-MB-231 cells with the ChIP assay kit (Milipore, Billerica, MA) following the manufacturer's protocol. After curcumin treatment, the cells were treated with 1% formaldehyde for 10 min followed by sonication. Immunoprecipitation was performed with control IgG and anti-p53 antibodies (2 µg). The region containing a putative p53-binding site on human Notch1 promoter was amplified by PCR with specific primers (273 bp): 5'-GACTCTGAGCCTCACTAGTGCC-3' and 5'-CTCCCCACCGGCTGCC CTCT-3'.
Terminal deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay
Apoptotic cells were confirmed with the DeadEndTM Fluorometric TUNEL System (Promega, Madison, WI), in accordance with the manufacturer's instructions. Exponentially growing cells were treated with curcumin for the indicated times or transfected with the indicated siRNAs or expression vectors. At 24 h after transfection, cells were treated with curcumin for the indicated times, fixed in 4% paraformaldehyde for 25 min at 4℃, and then permiabilized with 0.2% Triton X-100 for 5 min at room temperature. Free 3' ends of fragmented DNA were enzymatically labeled with the TdT-mediated dUTP nick end labeling (TUNEL) reaction mixture for 60 min at 37℃ in a humidified chamber. Labeled DNA fragments were monitored by fluorescence microscopy (Nikon, Instech Co., Ltd., Kanagawa, Japan).
RESULTS
Notch1 promoter contains a p53-binding site that functions as a curcumin-response element
To investigate whether Notch1 expression is affected by curcumin, the cells were treated with different concentrations of curcumin for 24 h, and Notch gene expression was measured by RT-PCR. Treatment of MDA-MB-231 cells with curcumin caused a significant reduction in Notch1 mRNA level and a slight reduction in Notch4 mRNA level, whereas the levels of Notch2 and Notch3 mRNA remained unchanged (Fig. 1A). In addition, curcumin significantly decreased the levels of total Notch1 protein (t-Notch) and cleaved Notch protein (c-Notch) in MDA-MB-231 cells in a dose- and time-dependent manner compared with the results obtained in the untreated control (Fig. 1B). We subsequently examined the expression pattern of Hes1, one of the well-known Notch1 target genes and found the level of Hes1 protein was reduced by curcumin (Fig. 1B). We next constructed a human Notch1 promoter-driven luciferase reporter plasmid, p(3286)luc (Supplementary Fig. 1A) and performed a reporter assay. The luciferase activity of p(3286)luc was significantly decreased by curcumin in a dose-dependent manner (Supplementary Fig. 1B). The pGL3 control vector without the Notch1 promoter sequence was unaffected by curcumin (Supplementary Fig. 1B), thus indicating that the cloned promoter region is sufficient for responding to curcumin. We next constructed luciferase reporter vectors named p(2366)luc, p(1273)luc, and p(484)luc, respectively (Fig. 1C). The reporter activities of the 3 constructs were repressed by curcumin (Fig. 1C), suggesting that the curcumin response element(s) might be located within the 484 bp fragment of the Notch1 promoter. We analyzed the sequence of the cloned 484 bp Notch1 promoter region and found one putative p53-binding site spanning from -264 to -228 bp relative to the translation initiation site in the human Notch1 promoter (Supplementary Fig. 1A). To examine whether p53 can directly bind to the putative p53-binding site in the Notch1 promoter, we performed EMSA by using a biotin-labeled oligonucleotide probe spanning the p53-binding sequence in the Notch1 promoter. As shown in Fig. 1E, a clear and strong protein-probe complex was observed in the nuclear extracts from MDA-MB-231 cells not treated with curcumin (lanes 2 and 5), whereas this complex was disrupted by curcumin treatment (lane 3). Specificity of the binding was validated by competition with an excess of unlabeled probe (lanes 6 and 7). We next investigated whether p53 binds to the Notch1 promoter in vivo by using ChIP assay with specific antibodies against p53 on the chromatin fragments from MDA-MB-231 cells (Fig. 1F). ChIP results showed that the association between p53 and the p53-binding site on the endogenous Notch1 promoter was detected in untreated cells. However, this binding was decreased in response to curcumin treatment (Fig. 1F).
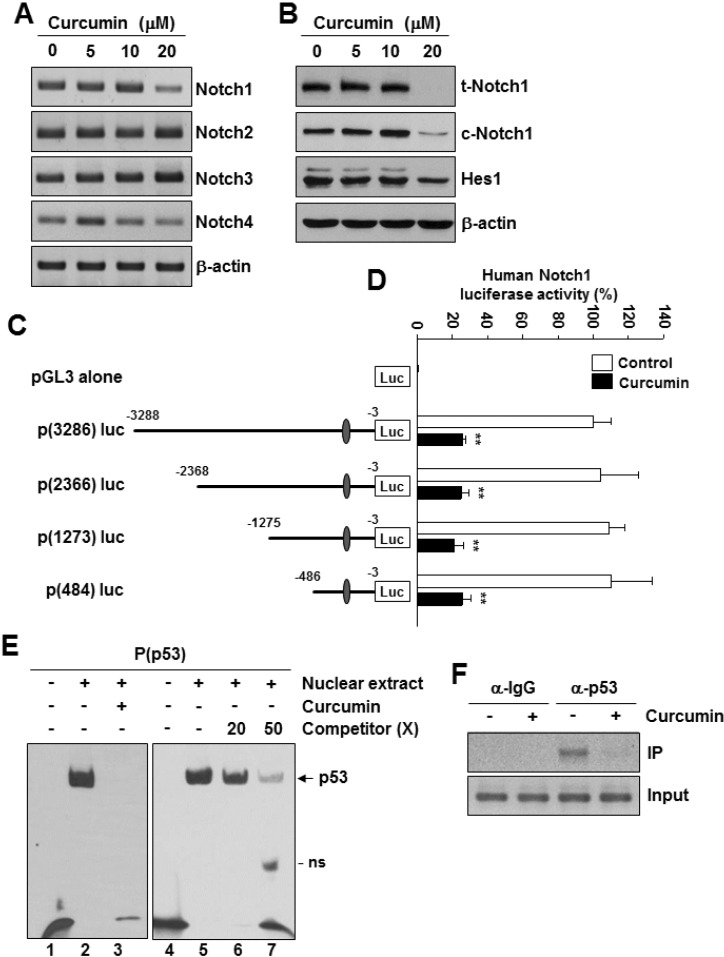
Fig. 1
Localization and identification of a 53-binding site in human Notch1 promoter. MDA-MB-231 cells were treated with different concentrations of curcumin (0~20 µM) for 24 h. (A) The mRNA levels of human Notch1, Notch2, Notch3, Notch4, and β-actin were detected by RT-PCR analysis. (B) The protein levels of Notch1, c-Notch1, Hes1, and β-actin were confirmed by Western blot analysis using the specific antibodies. (C) A schematic representation showing the deletion constructs of the human Notch1 promoter. (D) MDA-MB-231 cells were transfected with the indicated luciferase reporter vectors and subsequently treated with curcumin (20 µM) for 24 h. Cell extracts were prepared and analyzed for luciferase activity. **p<0.001 vs control, n=3. (E) The p53-binding activity in nuclear extracts was measured by EMSA. MDA-MB-231 cells were incubated with curcumin (20 µM) for 24 h. Nuclear extracts from MDA-MB-231 cells were incubated with biotinlabeled P(p53) oligonucleotide as a Notch1 promoter-specific p53 probe. In competition assay, 20x and 50x excess unlabeled probe were added to the reaction mixture. ns: nonspecific band. (F) MDA-MB-231 cells were treated with or without curcumin (20 µM) for 24 h. ChIP analysis was performed with control IgG or anti-p53 antibodies. IP, immunoprecipitation.
![]()
Notch1 expression is downregulated by p53 silencing
To examine the role of mutant p53 in Notch gene expression, we used siRNA targeting p53 to knockdown p53 expression. Both RT-PCR and western blot analyses showed reductions in p53 mRNA (Fig. 2A and Supplementary Fig. 2A) and protein (Fig. 2B) levels, respectively, in p53 siRNA-transfected cells. Among the 4 Notch genes, only Notch1 was downregulated by p53 knockdown at both mRNA (Fig. 2A and Supplementary Fig. 2A) and protein levels (Fig. 2B and Supplementary Fig. 2B). The cells transfected with p53 siRNA had lower levels of Hes1 mRNA and protein than the control siRNA-transfected cells (Fig. 2A and B). To confirm the involvement of p53 in regulation of Notch1 gene expression, we analyzed the activity of the Notch1 promoter with p53 silencing or overexpression. In MDA-MB-231 cells cotransfected with p(484)luc and p53 siRNA, the luciferase activity decreased by ~40% compared with that in control cells (Fig. 2C). In addition, while the overexpression of wild-type p53 had a suppressive effect on Notch1 promoter activity, overexpression of mutant p53 induced the activation of Notch1 promoter (Fig. 2D).
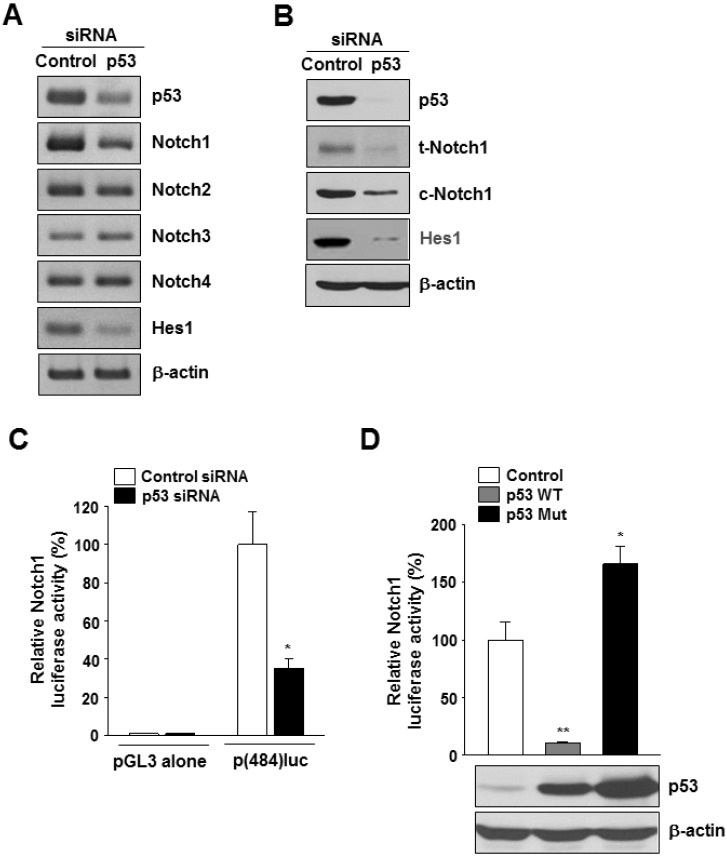
Fig. 2
Effects of p53 silencing on Notch1 expression. MDA-MB-231 cells were transfected with control siRNA or with p53 siRNA for 48 h. (A) The mRNA levels of human p53, Notch1, Notch2, Notch3, Notch4, Hes1, and β-actin were detected by RT-PCR analysis; representative RT-PCR. (B) The protein levels of human p53, total Notch1 (t-Notch1), cleaved Notch1 (c-Notch1), Hes1, and β-actin were detected by Western blot analysis; representative Western blot. (C) MDA-MB-231 cells were cotransfected with p(484)luc vector together with the indicated siRNAs, followed 48 h later by luciferase activity assay. *p<0.01 vs control siRNA, n=3. (D) MDA-MB-231 cells were cotransfected with p(484)luc together with a mammalian expression vector encoding wild-type p53 (p53WT) or mutant p53 (p53 Mut), or with control vector (pCMV-neo). After 48 h of transfection, cell extracts were prepared and analyzed for luciferase activity. **p<0.001; *p<0.01 vs control vector, n=3.
![]()
Curcumin-induced apoptosis is modulated by the p53-Notch1 axis
To investigate whether the p53-Notch1 axis plays a role in curcumin-induced cell apoptosis, we knocked down mutant p53 and Notch1 in MDA-MB-231 cells by siRNAs. Downregulation of p53 or Notch1 was confirmed at both mRNA and protein levels (Fig. 2A and B, and Supplementary Fig. 3). Then, the cells were treated with 20 µM curcumin for 48 h. As shown in Fig. 3, Notch1 siRNA- transfected MDA-MB-231 cells were more sensitive to spontaneous and curcumin-induced apoptosis than that of control cells, which was similar to p53 silencing. Next, we examined whether active Notch1 levels can block curcumin-induced apoptosis. There was a significant reduction in TUNEL-positive nuclear staining intensity among curcumin/Notch1 intracellular domain (NICD)-overexpressing cells compared with those treated with curcumin alone (Fig. 4A). Moreover, the number of TUNEL-positive apoptotic cells was reduced upon myc-tagged NICD overexpression compared with that of curcumin treatment (Fig. 4B). In addition, consistent with this observation, we observed a reduction in the levels of cleaved PARP and Caspase-3, and an increase in Bcl-2 level in accordance with the overexpressed level of exogenous NICD proteins (Fig. 4C).
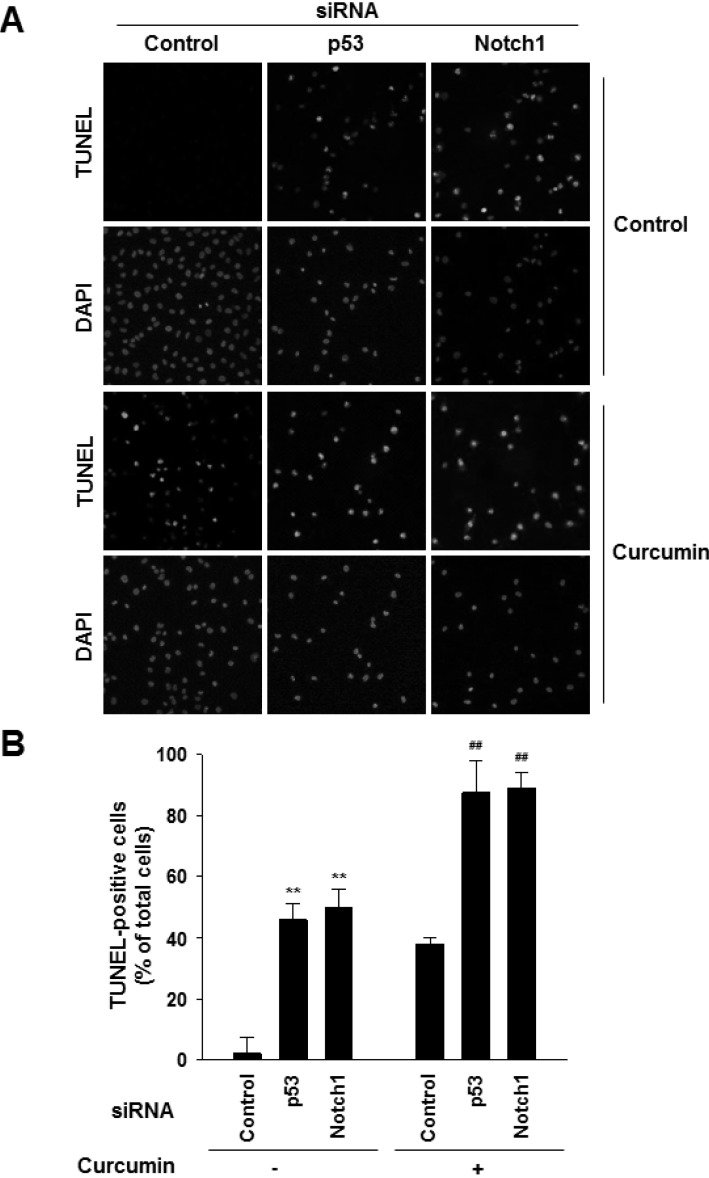
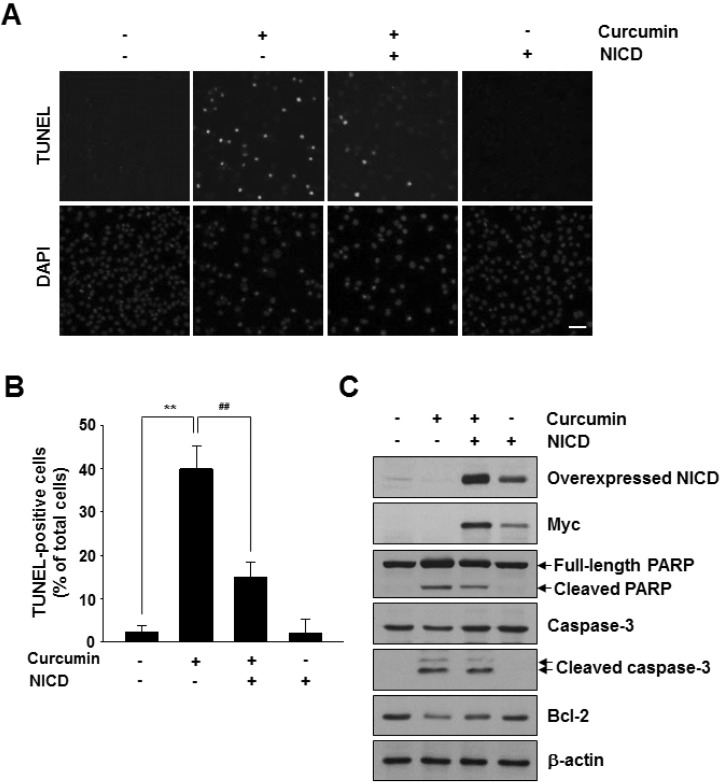
Fig. 3
Effects of down-regulation of mutant p53-Notch1 axis on curcumin-induced apoptosis. MDA-MB-231 cells were transfected with control siRNA or Notch1 siRNA. After 24 h of transfection, cells were treated with curcumin (20 µM) and incubated for 24 h. (A) Apoptosis was detected by TUNEL assay. DAPI stains nuclear DNA. Scale bar: 50 µm. (B) Values are expressed as the percentage of TUNEL-positive cells divided by total DAPI-positive cells counted. **p<0.001 vs control siRNA without curcumin, ##p<0.001 vs control siRNA with curcumin, n=3.
![]()
Fig. 4
Effects of overexpression of active Notch1 on curcumininduced apoptosis. MDA-MB-231 cells were transfected with an empty vector control or a mammalian expression vector encoding Myc-tagged NICD. After 24 h of transfection, cells were treated with curcumin (20 µM) and incubated for 48 h. (A) Apoptosis was detected by the TUNEL assay. DAPI stains nuclear DNA. Scale bar: 50 µm. (B) Values are expressed as the percentage of TUNELpositive cells divided by total DAPI-positive cells counted. *p<0.01; **p<0.001 vs control without curcumin, #p<0.01; ##p< 0.001 vs control with curcumin n=3. (C) Western blot analysis of Myc, NICD, PARP, Caspase-3, and Bcl-2 protein levels in NICD-transfected cells in the presence or absence of curcumin (20 µM).
![]()
DISCUSSION
Recent studies have demonstrated that wild-type p53 is involved in Notch1 gene induction in normal human epithelial cells, including keratinocytes [20,21]. In addition, the ectopic expression or restoration of wild-type p53 function in human cancer cell lines, which are deficient in p53 function or express wild-type p53, upregulates the expression of Notch1 [22]. On the other hand, p53 has been reported to downregulate Notch1 expression in mouse thymoma cells and human mesothelial cells [23,24]. Thus, these results suggest a cell type-specific mechanism of wild-type p53 control of Notch1 gene expression. However, very little is known with regard to a relationship between mutant p53 and Notch1. In this study, we demonstrated that Notch1 is a direct target of mutant p53 (R280K) in MDA-MB-231 cells. Overexpression of mutant p53 increased Notch1 promoter activity, whereas the knockdown of mutant p53 level by siRNA led to reduced expression of Notch1 and its target gene Hes1. In addition, mutant p53 bound to p53-response element in the Notch promoter in vitro and in vivo, indicating that mutant p53 is positively involved in the regulation of Notch1 gene expression in MDA-MB-231 cells.
Previous studies have identified MAP2K3, ID4, and chemokines CXCL5, CXCL8, and CXCL12 as transcriptional targets of endogenous gain-of-function p53 mutants, including R175H, R273H, and R280K, which are frequently detected in human breast cancers [27-29]. At present, we do not know whether other mutant p53 proteins such as R273H and R175H have the ability to induce Notch1 gene expression. Thus, further studies are needed to investigate the effects of other mutant p53 proteins on Notch1 gene expression in breast cancer cells.
Our data showed in this study that Notch1 expression was negatively regulated by ectopic wild-type p53 overexpression in MDA-MB-231 cells. This is in disagreement with previous reports showing the role of wild-type p53 as a positive regulator of Notch1 gene expression in MCF7 human breast cancer cells [22]. In the context of a mutant p53 (R280K)-expressing MDA-MB-231 cells, ectopic expression of wild-type p53 is unlikely to be sufficient to drive Notch1 transcription. Considering that mutant p53 acquires dominant-negative activity via its hetero-oligomerization with wild-type p53 [17-19], our data can be explained by the possibility that exogenous wild-type p53 proteins may bind to endogenous mutant p53, but forms nonfunctional oligomers that are unable to induce Notch1 gene expression, although the Notch1 gene is likely to be a common target of wild-type p53 and mutant p53 (R280K).
Some of endogenous p53 mutants have additional properties (gain-of-function), contributing to drug resistance and a higher rate of tumor growth and metastasis [18,19,30]. Among these p53 mutations, p53 mutant R280K is highly expressed in triple-negative breast cancer cell line, MDA-MB-231 cells [25,26]. Because patients with triple-negative breast cancers (negative for estrogen receptor, progesterone receptor, and HER2 expression: TNBC) typically have a poor prognosis [31], there is an increasing need for novel therapeutic approach that target TNBC. Our previous study showed that curcumin induced the apoptosis of MDA-MB-231 human breast carcinoma cells. It has been reported that mutant p53 mediates the survival of breast cancer cells [32] and that the downregulation of Notch1 expression regulates cell death through apoptosis [33]. Therefore, it is likely that mutant p53-Notch1 axis may have a close relationship with curcumin-induced apoptosis in MDA-MB-231 cells. Supporting this idea, in this study, we found that the knockdown of endogenous mutant p53 mutant (R280K) or Notch1 by siRNA induced apoptosis of MDA-MB-231 cells, which effects were further enhanced when combined with curcumin, whereas the ectopic expression of active Notch1 can rescue the survival defect induced by curcumin, suggesting that curcumin-induced apoptosis in MDA-MB-231 cells could be a consequence of a suppression of mutant p53-Notch1 axis. Thus, the targeting of mutant p53 and/or Notch1 may be combined with a chemotherapeutic strategy to improve the response of breast cancer cells to therapeutic agents such as curcumin.
We report, for the first time, that Notch1 suppression by curcumin is due to, at least in part, a reduction in the mutant p53 level, which may contribute to the curcumin-induced apoptotic response in vitro. Further in vivo studies will be needed to determine whether the combination of p53 or Notch1 siRNA and curcumin can be a possible therapeutic approach for better management of human breast cancer.
 XML Download
XML Download