

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Brain ischemia is caused by an interruption of cerebral blood flow that leads to stress, cell death, and inflammation and ischemic stroke is the second most common cause of death in the developed countries and causes severe disability [1]. Severe brain ischemia blocks the brain's energy supply, leading to mitochondrial dysfunction through oxygen and glucose deprivation [2,3]. The loss of oxygen blocks the oxidation of pyruvate by mitochondria, drastically reducing ATP production [3].
The sodium-potassium adenosine triphosphatase (Na-K ATPase) is an electrogenic transmembrane enzyme located in the plasma membrane of all animal cells and catalyzes the active transport of sodium ions out and potassium ions into the cells [4]. Na-K ATPase is responsible for the establishment of ion gradients necessary for cell function, including the maintenance of the resting membrane potential of neurons [5]. The Na-K ATPase consists of α and β subunits [6,7]. The catalytic α subunit contains the sites for binding Na+, K+ and ATP, and the β subunit is required for targeting the α subunit to the plasma membrane and for the occlusion of potassium ions [7,8]. Among α subunit isoforms, α1 and α2 subunits are strongly expressed in glial cells, while α1 and α3 subunits are expressed in neurons [9].
The brain is composed of not only neurons but also glial cells; more than 90% of the total cell population in the human brain is glial cells [10]. It is well known that astrocytes have critical functions in the brain, including maintenance of ionic homeostasis and prevention of excitotoxicity [3, 11-13]. The role of astrocytes in brain ischemia has been suggested to be a protective effect against ischemia-induced neuronal dysfunction [3,12]. However, the role of astrocytes in ischemic brain injury is still controversial.
Astrocytes maintain their energy through the operation of oxidative phosphorylation and glycolysis [14], and powerful Na-K pumps maintain Na+, K+, and Ca2+ gradients [15]. It has been suggested that the Na-K ATPase serves as a good marker of ischemic-reperfusion injury in neurons and glial cells [11]. Ischemia hyperpolarized the membrane potential of hippocampal astrocytes [16], activated the Na-K ATPase activity of cultured astrocytes [13], and enhanced the immunoreactivity of the Na-K ATPase α1 subunit in the gerbil hippocampus [17]. However, further studies are necessary to elucidate the role of astrocytic Na-K ATPase in the pathologic process of ischemic injury. Thus, we have examined the role of the Na-K ATPase in ischemic injury of primary cultured astrocytes.
Go to : 
METHODS
Primary astrocyte cultures
All experimental protocols were approved by the Institutional Animal Care and Use Committee of the Kyung Hee University and all efforts were made to minimize the number of animals utilized as well as animal suffering. Primary astrocyte culture was performed with slight modification of previously described methods [18]. One-day-old Sprague-Dawley rats were used. To identify astrocytes, cultured cells were immunocytochemically stained with antibodies to glial fibrillary acidic protein (GFAP) (Invitrogen, Carlsbad, CA), a specific marker for astrocytes. In this experiment, 90~95% of cultured cells were GFAP-positive (Fig. 1).
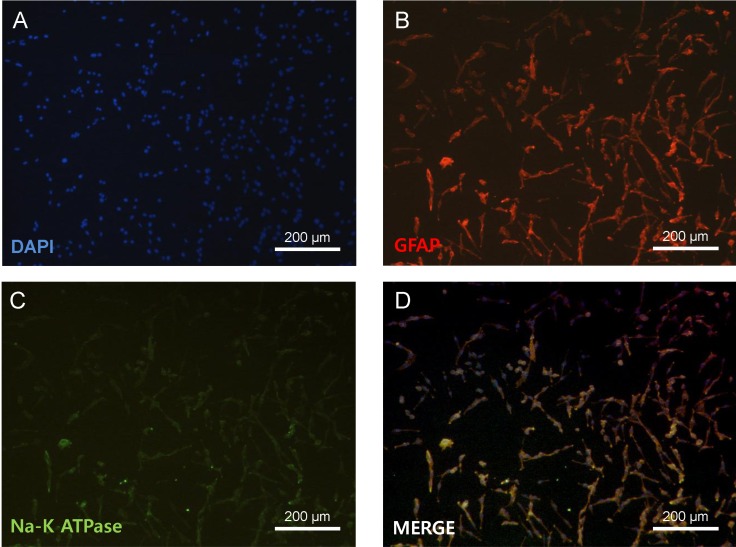
Induction of chemical ischemia in cultured astrocytes
Chemical ischemia was induced by a medium containing sodium azide (NaN3) without glucose to produce oxygenglucose deprivation conditions [19]. Primary astrocytes were seeded in 100 mm diameter dishes (5×106 cells per ml) or in 96-well culture plates (105 cells per ml). Cells were cultured in complete DMEM containing 10% fetal bovine serum and 1% penicillin/streptomycin. In chemical ischemia-treated groups, the glucose-free medium containing 3 mmol/l NaN3 and 10 mmol/l 2-deoxyglucose was applied, and the cells were incubated for 1, 2, 3, and 6 h at 37℃ in a humidified 5% CO2 atmosphere. In control groups, the medium containing glucose without NaN3 was applied and cells were incubated for 1, 2, 3, and 6 h in the same conditions.
MTT Assay for cell viability
MTT [3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide, Duchefa Biochemie, Haarlem, The Netherlands] is converted to the blue formazan product by metabolically active mitochondria, and the reaction is proportional to the number of viable cells [20,21]. Astrocytes were cultured in 96-well culture plates for 24 h and chemical ischemia was induced for 1, 2, 3, and 6 h. MTT (0.5 mg/ml) was added to the control and the ischemia-treated wells of the culture plate. After the MTT reaction ran for 2 h, the media in each well was aspirated and 100 µl of dimethyl sulfoxide was added to solubilize the blue crystals. The optical density was read with an ELISA plate reader at a wavelength of 570 nm.
Lactic dehydrogenase (LDH) assay for cell Cytotoxicity
Cytotoxicity is classically evaluated by the quantification of plasma membrane damage. The LDH-Cytotoxicity Assay Kit (Cytoscan™ LDH-cytotoxicity Assay kit, bio-WORLD Co., Dublin, OH) is a useful method for quantifying cytotoxicity based on the measurement of LDH release activity [20]. Astrocytes were cultured in a 96-well plate for 24 h and chemical ischemia was induced for 1, 2, 3, and 6 h. Aliquots of 50 µl in the control and the ischemia-treated wells were transferred to a new 96-well culture plate and were mixed with the LDH assay kit reagent for 20 min. A stop solution of 50 µl was added to each well and the optical density was read with an ELISA plate reader at a wavelength of 490 nm.
RNA isolation and real-time PCR
Total RNA was extracted and purified from primary cultured astrocytes using TRIZOL® Reagent (Invitrogen, Carlsbad, CA) [22]. cDNAs were generated using Super-Script™ II Reverse Transcriptase (Invitrogen, Carlsbad, CA) with a mixture of oligo(dt) (15 primer, Promega, Madison, WI) and 10 mmol/l dNTP (Real Biotech Co., Taipei, Taiwan). Real-time PCR was performed using a Light Cycler (Roche Diagnostics Ltd) and Light Cycler-Fast Start DNA Master SYBR Green I mix (Roche Diagnostics Ltd, Lewes, UK). The thermal cycling profile was as follows: preincubation step at 95℃ for 10 min, 50 cycle denaturation step at 95℃ for 10 s, annealing step at 62℃ for 8 s, and extension step at 72℃ for 15 s. The primer sequences were as follows: 5'-TGTGATTCTGGCTGAGAACG-3' (forward) and 5'-TCTTGCAGATGACCAAGTCE-3' (reverse) for the Na-K ATPase α1 subunit (Gene Bank™ Accession No. NM_144900). The β-actin primers were as follows: 5'-ATGGGTCAGAAGGACTCCTACG-3' (forward) and 5'-ATGGGTACGACCAGAGGCATAC-3' (reverse).
Membrane fraction from cultured astrocytes
Preparation of the total membrane fraction was performed as described previously [23]. Briefly, cultured astrocytes in 100 mm culture dishes were washed and scraped in 5 ml of PBS. After centrifugation at 450 g for 5 min, pellets were re-suspended in a culture dish with cold homogenization buffer (250 mmol/l sucrose, 2 mmol/l EGTA, 5 mmol/l sodium azide, 20 mmol/l HEPES, pH 7.4) containing freshly added protease inhibitors (Complete, Mini, EDTA-free, Roche Applied Science, Mannheim, Germany). The homogenate was centrifuged at 450 g for 5 min, and the supernatant (SN1) was kept. This procedure was repeated and the next supernatant (SN2) was collected. The supernatant was centrifuged at 190,000 g for 70 min, and the pellet was collected as the total membrane fraction.
Western blotting
A total of 30 µg of protein prepared from the control and the ischemia-treated astrocytes was separated on 10% SDS-PAGE gels and transferred onto nitrocellulose membranes (Bio-Rad, Hercules, CA). The transferred membranes were incubated in blocking buffer (25 mmol/l Tris-Cl, 150 mmol/l NaCl, 0.05% Tween-20, and 5 g/100 ml non-fat dry milk, pH 8.0) at room temperature for 1 h and washed three times with TBST (25 mmol/l Tris-Cl, 150 mmol/l NaCl, 0.05% Tween-20, pH 8.0). The transferred membranes were incubated overnight at 4℃ with primary antibodies including anti-α1 Na-K ATPase (1:5,000, mouse monoclonal; Abcam, San Francisco, CA) and anti-β-actin (1:2,000, mouse monoclonal; Santa Cruz Biotechnology, Paso Robles, CA). After incubation with primary antibodies, the membranes were washed three times with TBST and incubated in horseradish peroxidase-conjugated goat anti-mouse IgG antibody (1:10,000 or 1:4,000; Stressgen, Ann Arbor, MI) at room temperature for 1 h. The membranes were washed three times with TBST and protein bands were visualized with an enhanced chemiluminescence reagent (SuperSignal West Pico Chemiluminescent Substrate, Thermo Fisher Scientific, Waltham, MA). Western blotting was performed on samples from more than three different cultures.
Assay of Na-K ATPase specific activity
Na-K ATPase activity of the control and the ischemia-treated astrocytes was measured as described previously [24]. Preparation of the total membrane fraction from the control and the ischemia-treated astrocytes was performed as described above. Reaction mixtures for Na-K ATPase activity assay was as follows: (i) 30 mmol/l imidazole-HCl, 130 mmol/l NaCl, 20 mmol/l KCl, 4 mmol/l MgCl2 and (ii) 30 mmol/l imidazole-HCl, 4 mmol/l MgCl2, and 1 mmol/l ouabain at pH 7.4. The reaction was started by adding 4 mmol/l Tris-ATP and incubated at 37℃ for 10 min. The enzymatic hydrolysis of ATP was terminated by 100 µl of 12.5% ice-cold trichloroacetic acid. Enzymatic activity was measured as a function of liberated inorganic phosphate (Pi) by the colorimetric reaction. Enzyme activity was calculated as the difference in the Pi content in nmol/min between media (i) and (ii). Enzyme protein was solubilized using 100 µl of 20% SDS. From this mixture an aliquot (0.2 ml from a total volume of 1.1 ml) was taken for Pi estimation followed by quick addition of 0.8 ml of reagent A (containing 3% ascorbic acid in 0.5 N HCl and 0.5% ammonium molybdate solution) in a total volume of 1 ml and the tubes were kept on ice for 10 min. Then, 1 ml of reagent B (containing 2% sodium meta-arsenite, 2% trisodium citrate, and 2% acetic acid) was added. The color developed after 10 min at 37℃ and was read at a wavelength 850 nm with a spectrophotometer (SpectraMax 190, Molecular Devices, Sunnyvale, CA).
Statistical analysis and Quantification of PCR and Western blots
Data are presented as the mean±S.E.M. The Student's t-test was used for statistical analyses. p values less than 0.05 were considered significant. To quantify mRNA and protein in real-time PCR and Western blot experiments, the background intensity was first subtracted using ImageJ software (developed at the U.S. National Institutes of Health and available at http://rsb.info.nih.gov/ij/).
Go to :
RESULTS
Effect of chemical ischemia on the cytotoxicity and viability of cultured astrocytes
To examine the effect of duration of chemical ischemia on the cytotoxicity and viability of astrocytes, we measured LDH release and MTT conversion at 1, 2, 3, and 6 h after the induction of chemical ischemia in cultured astrocytes (Fig. 2). At each time, we compared the cytotoxicity and viability of ischemia-treated astrocytes to those of control cells. In the LDH assay, cytotoxicity of cultured astrocytes was not altered at 1 h of chemical ischemia (107±5% of control, p>0.05) and was significantly increased at 2, 3, and 6 h (152±21%, 202±23%, and 226±30% of time-matched controls; p<0.05, p<0.01, and p<0.001, respectively; data were obtained from five experiments; Fig. 2A). The MTT assay showed that viability of cultured astrocytes was significantly reduced at 1, 2, 3, and 6 h of chemical ischemia (80±2%, 69±3%, 56±3%, and 51±30% of time-matched controls; p<0.001; data were obtained from five experiments; Fig. 2B).
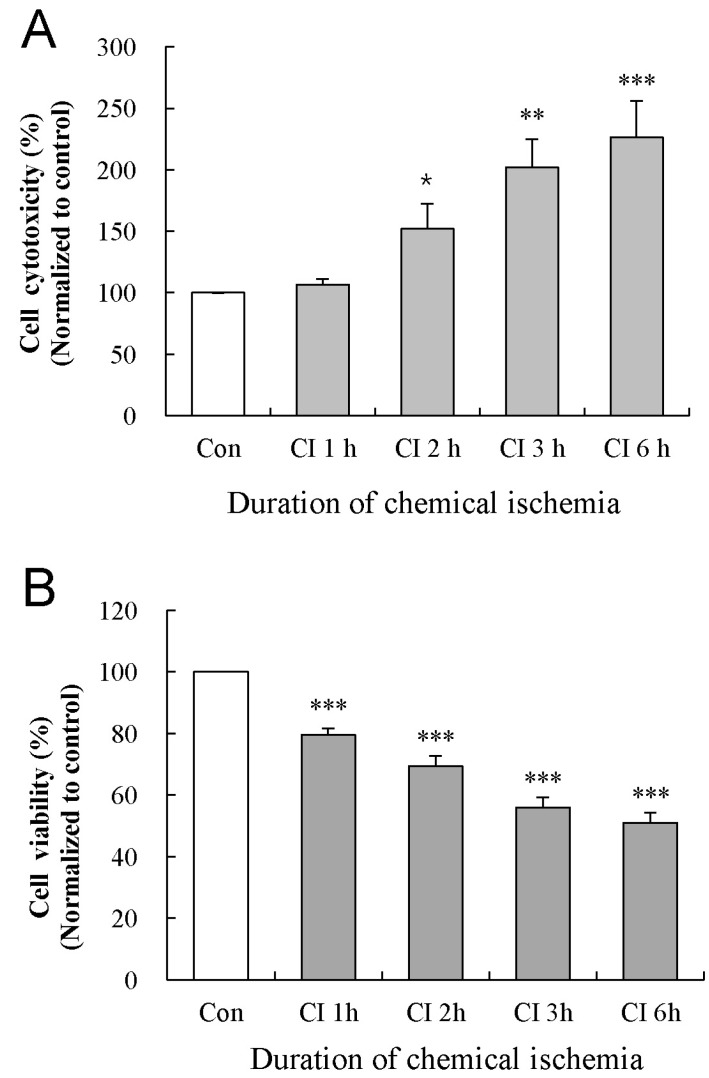 | Fig. 2Effect of chemical ischemia on the cytotoxicity and viability of cultured astrocytes. Cytotoxicity and cell viability was measured by LDH and MTT assays, respectively. The levels of LDH release (A) and MTT reduction (B) were quantified and compared to the control at each time point. Each value indicates the mean±S.E.M. normalized to the control of each time point. Data were obtained from five experiments. *p<0.05, **p<0.01, and ***p<0.001.
|
Effects of chemical ischemia on the protein and mRNA expression of the Na-K ATPase α1 subunit in cultured astrocytes
Western blotting was performed to elucidate the changes in the protein expression of the Na-K ATPase α1 subunit in cultured astrocytes following chemical ischemia for 1, 2, 3, and 6 h (Fig. 3Aa & b). Protein expression of the Na-K ATPase α1 subunit was significantly increased to 163±27% of control at 2 h of chemical ischemia (p<0.05; data were obtained from five experiments) and returned to control levels at 3 h of chemical ischemia (113±20% of the control, p>0.05). At 6 h of chemical ischemia, protein expression was significantly decreased to 63±14% of control (p<0.05).
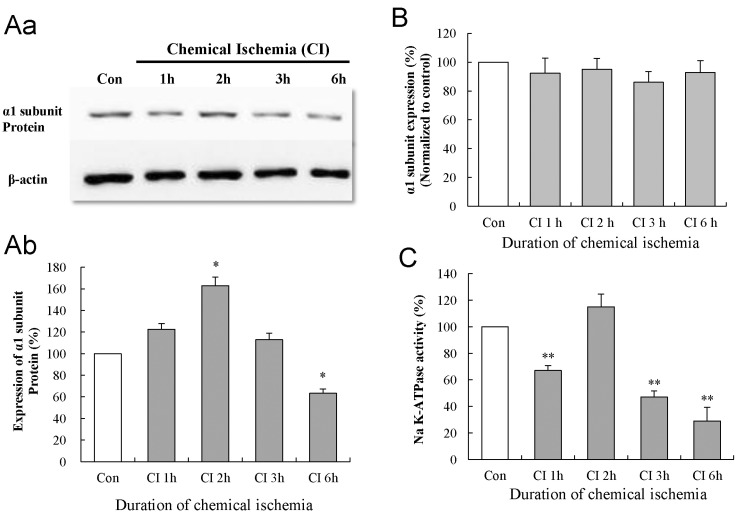 | Fig. 3Effect of chemical ischemia on the expression of Na-K ATPase α1 subunit and Na-K ATPase activity in cultured astrocytes. (A) Western blot analysis shows the expression of Na-K ATPase α1 subunit protein following chemical ischemia for 1, 2, 3, and 6 h. Data were obtained from five experiments. (B) Real-time PCR analysis shows the expression of Na-K ATPase α1 subunit mRNA in cultured astrocytes following chemical ischemia for 1, 2, 3, and 6 h. Data were obtained from four experiments. (C) The activity of Na-K ATPase in the membrane fraction of cultured astrocytes after chemical ischemia for 1, 2, 3, and 6 h. Data were obtained from five experiments. For all experiments, each value indicates the mean±S.E.M. normalized to the control of each time point. *p<0.05, **p<0.01.
|
Real-time PCR was performed to examine changes in the
expression of Na-K ATPase α1 subunit mRNA in cultured astrocytes following chemical ischemia for 1, 2, 3, and 6 h (Fig. 3B). Chemical ischemia for 1, 2, 3, and 6 h did not change the expression of α1 subunit mRNA (p>0.05; data were obtained from nine experiments).
Effects of chemical ischemia on Na-K ATPase activity in cultured astrocytes
To elucidate the relationship between the change in expression of the Na-K ATPase α1 subunit and the activity of the Na-K ATPase, we measured Na-K ATPase activity in the membrane fraction of cultured astrocytes following chemical ischemia for 1, 2, 3, and 6 h (Fig. 3C). In this experiment, Na-K ATPase activity of the control groups was 18.4±4.5 nmol Pi/µg protein per min. Chemical ischemia-treated groups showed a gradual and progressive decrease in Na-K ATPase activity to 67±4%, 47±5%, 29±10% of control at 1, 3, and 6 h of chemical ischemia, respectively (p<0.01; data were obtained from five experiments). However, at 2 h of chemical ischemia, Na-K ATPase activity recovered and was not significantly different from the control level (116±10% of the control, p>0.05).
Effect of reoxygenation on chemical ischemia-induced cytotoxicity of cultured astrocytes
To examine the effect of reoxygenation on chemical ischemia-induced cytotoxicity, we measured LDH release and MTT conversion after reoxygenation for 24 h following the induction of chemical ischemia for 1, 2, 3, and 6 h in cultured astrocytes (Fig. 4A and B). At each time, we compared the cytotoxicity and viability of ischemia/reoxygenation-treated astrocytes to those of time-matched control cells. After the induction of chemical ischemia for 1, 2, 3, and 6 h, the medium was changed to the control medium containing glucose without NaN3, and the cells were incubated at 37℃ for 24 h in humidified 5% CO2 atmosphere. In the LDH assay, cytotoxicity of cultured astrocytes was not altered at 1 and 2 h of chemical ischemia (95±2% and 122±18% of control, respectively; p>0.05) but was significantly increased at 3 and 6 h (182±17% and 200±22% of control, respectively; p<0.01; data were obtained from five experiments; Fig. 4A). In the MTT assay, viability of cultured astrocytes was significantly reduced to 85±4%, 72±5%, 57±11%, and 55±16% of control at 1, 2, 3, and 6 h of chemical ischemia, respectively (p<0.01, p<0.001, p<0.01, p<0.05, respectively; data were obtained from five experiments; Fig. 4B).
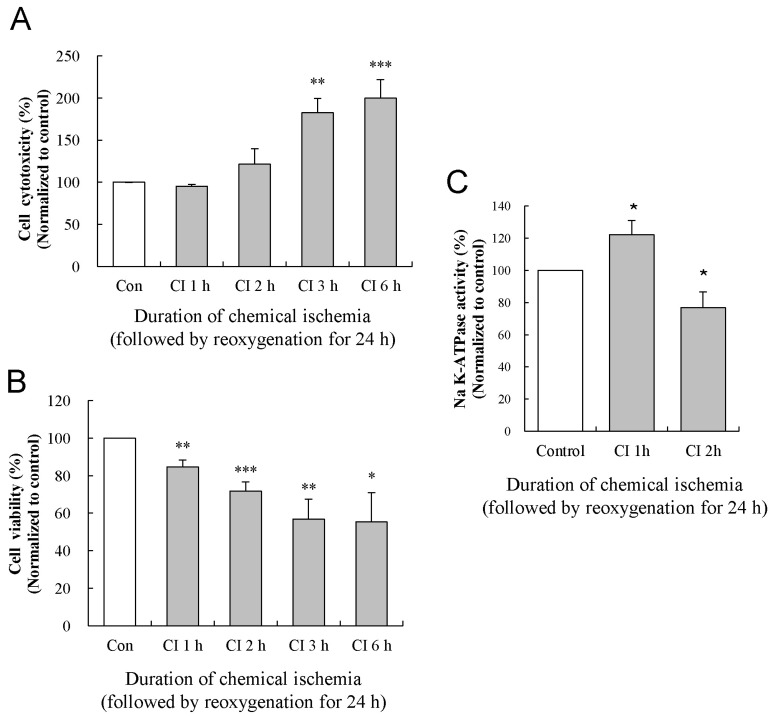 | Fig. 4Effects of reoxygenation on the changes in the cytotoxicity, viability, and activity of the Na-K ATPase. The levels of LDH release (A) and MTT reduction (B) were quantified at 24 h of reoxygenation following chemical ischemia for 1, 2, 3, and 6 h. (C) Activity of Na-K ATPase was measured at 24 h of reoxygenation following chemical ischemia for 1 and 2 h. Data show the mean± S.E.M. of the relative values obtained from five animals. *p<0.05, **p<0.01, and ***p<0.001.
|
Effect of reoxygenation on the alteration of Na-K ATPase activity induced by chemical ischemia
We next investigated a possible role of increased Na-K ATPase expression and activity in the effect of reoxygenation on ischemic cytotoxicity at 2 h of chemical ischemia. We examined the effect of reoxygenation on Na-K ATPase activity at 1 and 2 h of chemical ischemia. Reoxygenation following chemical ischemia for 1 h significantly increased the Na-K ATPase activity (122±9% of control; p<0.05), while reoxygenation following chemical ischemia for 2 h significantly decreased the enzyme activity (77±10% of control; p<0.05; data were obtained from seven experiments; Fig. 4C). Although Na-K ATPase activity at 3 and 6 h of chemical ischemia followed by reoxygenation was very difficult to measure due to the decreased cell viability, Na-K ATPase activity showed a decreasing tendency similar to the results measured after chemical ischemia alone (data not shown).
Go to :
DISCUSSION
Previous reports have suggested the involvement of astrocytes in ischemic neuronal injury [3,12,16]. In addition, some investigators have examined the role of astrocytic Na-K ATPase in ischemic injury [11,15]. However, the role of the Na-K ATPase in ischemic injury of astrocytes is unclear. In the present study, chemical ischemia-induced cytotoxicity of cultured astrocytes began to appear at 2 h of ischemia (Fig. 2) and the cytotoxic effect at 2 h was significantly blocked by reoxygenation for 24 h following chemical ischemia (Fig. 4). These results suggest that the critical time for ischemia-induced cytotoxicity in astrocytes might be 2 h after the initiation of ischemic insult. However, the result of cytotoxicity assay measured by LDH release was not correlated with that of cell viability assay measured by MTT reduction (Fig. 2 and Fig. 4). Especially, 1 h of chemical ischemia did not show any cytotoxicity but significantly reduced cell viability. In contrast, 2 h of chemical ischemia showed a significant effect on both cytotoxicity and viability. Due to this discrepancy, we focused on the cytotoxicity as an indicator of ischemic injury and decided 2 h of chemical ischemia as the critical time of ischemic astrocytic injury.
The expression of Na-K ATPase α1 subunit protein was increased at 2 h of chemical ischemia and was decreased at 6 h of ischemia, whereas the expression of mRNA was not changed at 1~6 h of chemical ischemia (Fig. 3). A previous report performed in gerbil hippocampus showed a similar result-that the immunoreactivity of Na-K ATPase α1 subunit in glial cells was enhanced after ischemic insult- and suggested that the enhancement of Na-K ATPase α1 subunit immunoreactivity may be a compensatory response to regulate the ion homeostasis in the brain [17]. In the present study, the expression of Na-K ATPase α1 subunit mRNA was not changed. It is well known that a change in protein expression is not correlated to that of mRNA. A similar discrepancy was also shown in a study for Na-K ATPase expression during ischemia-reperfusion injury in rat kidney where ischemia-reperfusion injury increased the expression of Na-K ATPase α1 subunit protein in the renal cortex, but the expression of Na-K ATPase α1 subunit mRNA was not changed [25].
The Na-K ATPase is responsible for maintenance of the resting membrane potential [5], and the catalytic α subunit of Na-K ATPase contains the sites for binding of Na+, K+ and ATP [8]. Increased expression of Na-K ATPase α1 subunit protein may result in increased hyperpolarization of the membrane potential. A previous study showed that the hyperpolarization of astrocyte membrane potential was meditated by the activation of the Na-K ATPase during oxygen- and glucose-deprivation recovery. The study also suggested that astrocytes might rescue endangered neurons during acute ischemic insult via their various homeostatic functions [16]. In the present study, the activity of Na-K ATPase was time-dependently decreased at 1, 3, and 6 h of chemical ischemia, whereas the enzyme activity was temporarily recovered to the control value at 2 h (Fig. 3). These results indicate that the decreased activity of the Na-K ATPase during ischemia might be due to the decreased production of ATP. Additionally, the Na-K ATPase α1 subunit might be up-regulated at 2 h of ischemia to compensate the decreased Na-K ATPase activity and the initiation of ischemic cytotoxicity.
We found that reoxygenation following 1 h of chemical
ischemia significantly increased the activity of the Na-K ATPase, whereas the enzyme activity was slightly decreased by reoxygenation following 2 h of ischemia (Fig. 4). These results indicate that before the initiation of cytotoxicity at 1 h of chemical ischemia, reoxygenation could induce a compensatory increase in Na-K ATPase activity. In contrast, after initiation of cytotoxicity at 2 h of ischemia, reoxygenation could not increase or maintain the activity of Na-K ATPase at the level of 2 h of ischemia. Thus, these results also suggest that the decreased activity of the Na-K ATPase during ischemia might be due to the restriction of energy and oxygen supply and that the change in Na-K ATPase α1 subunit expression might be compensatory to the decreased Na-K ATPase activity. In conclusion, we suggest that the critical time for cytotoxicity in astrocytes might be 2 h after the initiation of ischemic insult and that the expression and activity of the Na-K ATPase might increase at this time to protect against the cytotoxic effect of chemical ischemia.
Go to :
 XML Download
XML Download