

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
AB
absolute bioavailability
AUC
area under the plasma concentration-time curve
BA
bioavailability
Cmax
peak plasma concentration
CLt
total body clearance
CYP
cytochrome P450
Kel
elimination rate constant
P-gp
P-glycoprotein
PK
pharmacokinetics
TBC
total body clearance
Tmax
time to reach peak plasma concentration
t1/2
half-life
RB
relative bioavailability
INTRODUCTION
Repaglinide
[(S)-(+)-2-ethoxy-4-(2-oxo-2-[(α-isobuty1-2-piperidinobenzyl) amino]ethyl)-benzoic acid, a carbamoylmethyl benzoic acid (CMBA) derivative] is a novel, fast-acting prandial oral hypoglycemic agent developed for the treatment of patients with type 2 diabetes whose disease can not be controlled by diet and exercise alone [1]. It can reduce the fasting glucose concentrations in patients with type 2 diabetes mellitus. It helps to control blood glucose by increasing the amount of insulin released by the pancreas [2]. Repaglinide stimulates the release of insulin from the pancreatic beta cells by binding to and closing ATP-dependent potassium channels. This depolarizes the plasma membrane, leading to the opening of voltage-dependent calcium channels. Influx of calcium ions, which increases intracellular Ca2+, triggers exocytosis of insulin [3].
Repaglinide is rapidly absorbed from the gastrointestinal tract after oral administration. It differs from other antidiabetic agents in its structure, binding profile, duration of action and mode of excretion [4]. Repaglinide is primarily metabolized via oxidative biotransformation through the hepatic microsomal cytochrome P450 system, particularly the CYP3A4 isoform [5,6]. The metabolic pathway of repaglinide involves two major sites for its principal biotransformation: the piperidine ring and the aromatic carboxylic acid group [7]. Repaglinide has affinity for P-glycoprotein (P-gp) and it can significantly contribute to potential drug-drug interactions with other P-gp substrates or inhibitors [5,6,8]. Kajosaari et al. reported that co-administration of repaglinide with the CYP3A4 and OTAP inhibitor, cyclosporine A and gemfibrozil which significantly increased the plasma concentrations of repaglinide in humans [6,9].
Fluvastatin is rapidly and completely absorbed from the gastrointestinal tract but undergoes extensive first-pass metabolism in the liver mainly by CYP 2C9 and CYP 3A4 [10,11]. Fluvastatin is a the synthetic HMG-CoA reductase inhibitor [12,13]. It is an antilipemic agent and is also used as an adjunct to dietary therapy to slow the progression of coronary atherosclerosis in hypercholesterolemic patients suffering from diabetic mellitus or coronary heart disease [12,13].
CYP3A4 inhibitors (erythromycin, ketoconazole and itraconazole) have no effect on fluvastatin pharmacokinetics, in contrast to HMG-CoA reductase inhibitors that are metabolized by CYP3A4 [14,15]. However, bi-directional pharmacokinetic interactions between fluvastatin and CYP3A4 substrates have not been evaluated and it is not clear yet whether fluvastatin can alter the pharmacokinetics of CYP3A4 substrates such as repaglinide. The inhibitory effect of fluvastatin against CYP enzymes and P-gp is ambiguous elsewhere. Therefore, we evaluated the inhibition of CYP3A4 activity and P-gp activity by fluvastatin using CYP inhibition assays and rhodamine-123 retention assays in P-gp-overexpressing MCF-7/ADR cells.
Furthermore, the present study aimed to investigate the effect of fluvastatin on the pharmacokinetics of repaglinide in rats. Antidiabetic agents are commonly co-administered with cholesterol-lowering agents in clinics.
There are few reports about the effects of fluvastatin on the bioavailability or pharmacokinetics of other drugs in rats [16]. Hypercholesterolemia can be induced by complications of diabetes and repaglinide and HMG-CoA Reductase Inhibitor can be prescribed clinically for treatment of diabetes or hypercholesterolemics, complications of diabetes [5,17]. However, the effect of fluvastatin on the pharmacokinetics of repaglinide in vivo has not yet been reported. Thus, the purpose of this study was to investigate the possible effects of fluvastatin on the CYP3A4 and P-gp activity and the bioavailability or pharmacokinetics of repaglinide after oral and intravenous administration of repaglinide with fluvastatin in rats.
Go to : 
METHODS
Materials
Repaglinide, indomethacin (internal standard) and fluvastatin were purchased from Sigma-Aldrich Co. (St. Louis, MO, USA). HPLC-grade methanol and acetonitrile were acquired from Merck Co. (Darmstadt, Germany). All other chemicals for this study were of reagent grade and were used without further purification. Apparatus used in this study included an HPLC equipped with a Waters 1515 isocratic HPLC Pump, a Waters 717 plus auto sampler and a Waters™ 2487 scanning UV detector (Waters Co., Milford, MA, USA), an HPLC column temperature controller (Phenomenex Inc., CA, USA), a Bransonic® Ultrasonic Cleaner (Branson Ultrasonic Co., Danbury, CT, USA), a vortex-mixer (Scientific Industries Co., NY, USA), a high-speed microcentrifuge (Hitachi Co., Tokyo, Japan), and the Onetouch® Ultra™ Blood Glucose Monitoring System (LifeScan Inc., CA, USA).
Animal experiments
Male Sprague-Dawley (SD) rats (weighing 270~300 g) were purchased from Dae Han Laboratory Animal Research Co. (Choongbuk, Korea) and were given access to a commercial rat chow diet (No. 322-7-1, Superfeed Co., Gangwon, Korea) and tap water. The animals were housed, two per cage, and maintained at 22±2℃ and 50~60% relative humidity under a 12:12 h light: dark cycle. The experiments were initiated after acclimation under these conditions for at least 1 week. The Animal Care Committee of Chosun University (Gwangju, Korea) approved the design and conduct of this study. The rats were fasted for at least 24 h prior to the experiments and each animal was anaesthetized lightly with ether. The left femoral artery and vein were cannulated using polyethylene tubing (SP45, i.d. 0.58 mm, o.d. 0.96 mm; Natsume Seisakusho Co. Ltd., Tokyo, Japan) for blood sampling and i.v. injection, respectively.
Drug administration
The rats were divided into six groups (n=6, each): oral group (0.5 mg/kg of repaglinide dissolved in distilled water, 1.0 ml/kg) without (control) or with 1 and 3 mg/kg of fluvastatin (mixed in distilled water, 3.0 ml/kg), and an i.v. control group (0.2 mg/kg of repaglinide, dissolved in 0.9% NaCl solution, 1.5 ml/kg) without or with 1 and 3 mg/kg of fluvastatin (mixed in distilled water, 3.0 ml/kg). Oral repaglinide was administered intragastrically using a feeding tube, and fluvastatin was administered in the same manner 30 min prior to the oral administration of repaglinide. Repaglinide for i.v. administration was injected through the femoral vein within 0.5 min. A 0.3-ml aliquot of blood was collected into heparinized tubes from the femoral artery at 0.25, 0.5, 0.75, 1, 2, 4, 6, and 10 h after repaglinide oral administration and at 0 (to serve as control), 0.1, 0.25, 0.5, 1, 2, 6, and 10 h after repaglinide i.v. administration. The blood samples were centrifuged at 13,000 rpm for 3 min, and the plasma samples were stored at -40℃ until HPLC analysis.
HPLC analysis
Plasma concentration of repaglinide was determined by HPLC as reported by Ruzilawati et al. [18] with a slight modification. Briefly, a 50-µl aliquot of 1 µg/ml indomethacin, as an internal standard, and a 0.2-ml aliquot of 0.1 mol/l potassium dihydrogen orthophosphate (KH2PO4, pH 5.9) were mixed with a 0.15-ml aliquot of the plasma sample. After the mixture was vortexed, 1 ml of ethylacetate, 50 µl of isoamylalcohol and 35 µl of 1.0 M NaOH were added. The resulting mixture was then vortex-mixed for 10 min and centrifuged at 9,000 rpm for 10 min. After centrifugation, the ethylacetate phase was transferred into a clean test tube and evaporated under a gentle stream of nitrogen gas at 45℃. The dried extract was reconstituted with 150 µl of mobile phase, vortex-mixed and transferred to a clean autosampler vial. A 70-µl aliquot of the supernatant was injected into the HPLC system. Chromatographic separation was achieved using a µBondapak™ C18 column (3.9×300 mm i.d., 10 µm, Waters Co.) attached to a µBondapak™ C18 guard column (3.9×20 mm i.d., 10 µm, Waters Co.). The mobile phase consisted of 20 mM dipotassium hydrogen phosphate (pH 2.7, adjusted with phosphoric acid):acetonitrile (60:40, v/v). The flow-rate of the mobile phase was maintained at 1.0 ml/min. Chromatography was performed at 30℃ and regulated by an HPLC column temperature controller. The UV detector was operated at a wavelength of 244 nm. Repaglinide and indomethacin were eluted with retention times of 7.4 and 8.4 min, respectively. The lower limit of quantification for repaglinide in rat plasma was 10 ng/ml. The intra- and inter-day coefficients of variation of repaglinide were below 12.0 and 11.5%, respectively.
Determination of blood glucose concentrations
Blood samples were collected after oral and intravenous repaglinide administration at 0, 1, 3, 6, and 10 h. Blood glucose concentrations were measured immediately using the Onetouch® Ultra™ Blood Glucose Monitoring System and the glucose assay kit from BioAssay Systems (QuantiChrom™ Glucose Assay Kit) as described by Yoon and Mekalanos [17].
CYP inhibition assay
The assays of inhibition of human CYP3A4 enzyme activity were performed in a multiwell plate using the CYP inhibition assay kit (GENTEST, Woburn, MA, USA) as described previously [19]. Briefly, human CYP enzymes were obtained from baculovirus-infected insect cells. CYP substrate (7-BFC) was incubated with or without fluvastatin in enzyme/substrate buffer consisting of 1 pM of P450 enzyme and a NADPH generating system (1.3 mM NADP, 3.54 mM glucose 6-phosphate, 0.4 U/ml glucose 6-phosphate dehydrogenase and 3.3 mM MgCl2) in potassium phosphate buffer (pH 7.4). Reactions were terminated by adding stop solution after 45-min incubation. Metabolite concentrations were measured by spectrofluorometer (Molecular Devices, Sunnyvale, CA, USA) at an excitation wavelength of 409 nm and an emission wavelength of 530 nm. Positive control (1 µM ketoconazole) was run on the same plate and produced 99% inhibition. Results are expressed as the percent of inhibition.
Rhodamine-123 retention assay
The procedures used for the Rho-123 retention assay were similar to a previously reported method [20]. MCF-7/ADR cells were seeded in 24-well plates and pre-incubated with fluvastatin and verapamil for 30 min. At 80% confluence, the cells were incubated in FBS-free DMEM for 18 h. The culture medium was changed to Hanks' balanced salt solution and the cells were incubated at 37℃ for 30 min. After incubation of the cells with 20 µM rhodamine-123 in the presence or absence of fluvastatin (50 and 100 µM) for 90 min, the medium was completely removed. The cells were then washed three times with ice-cold phosphate buffer (pH 7.0) and lysed in EBC lysis buffer. Rhodamine-123 fluorescence in the cell lysates was measured using excitation and emission wavelengths of 480 and 540 nm, respectively. Fluorescence values were normalized to the total protein content of each sample and are presented as the ratio to control.
Pharmacokinetic analysis
The plasma concentration data were analyzed by the non-compartmental method using WinNonlin software version 4.1 (Pharsight Co., Mountain View, CA, USA). The elimination rate constant (Kel) was calculated by log-linear regression of repaglinide concentration during the elimination phase. The terminal half-life (t1/2) was calculated by 0.693/Kel. The peak plasma concentration (Cmax) and time to reach peak plasma concentration (Tmax) of repaglinide in plasma were obtained by visual inspection of the data from the concentration-time curve. The area under the plasma concentration-time curve from 0 h to the time (AUC0-t) of last measured concentration (Clast) was calculated by the linear trapezoidal rule. The AUC from zero to infinite (AUC0-∞) was obtained by the addition of AUC0-t and the extrapolated area determined by Clast/Kel. Total body clearance (CL/F) was calculated by:
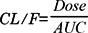
The absolute bioavailability (AB) was calculated by:
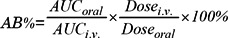
The relative bioavailability (RB) was calculated by AUCwith fluvastatin/AUCcontrol
* 100
Statistical analysis
Statistical analysis was conducted using one-way ANOVA followed by a posteriori testing with the Dunnett correction. Differences were considered to be significant at p<0.05. All mean values are presented with their standard deviation (Mean±SD).
Go to :
RESULTS
Inhibition of CYP3A4
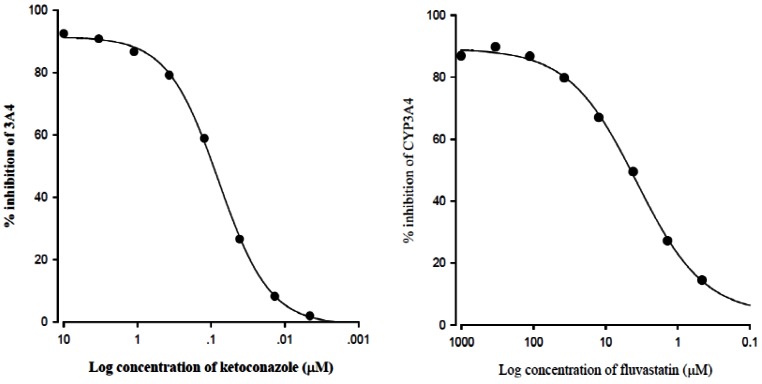
The inhibitory effect of fluvastatin on CYP3A4 activity is shown in Fig. 1. Fluvastatin inhibited CYP3A4 activity in a concentration-dependent manner. Fluvastatin strongly inhibited CYP3A4 with an IC50 value of 4.1 µM.
Rhodamine-123 retention assay
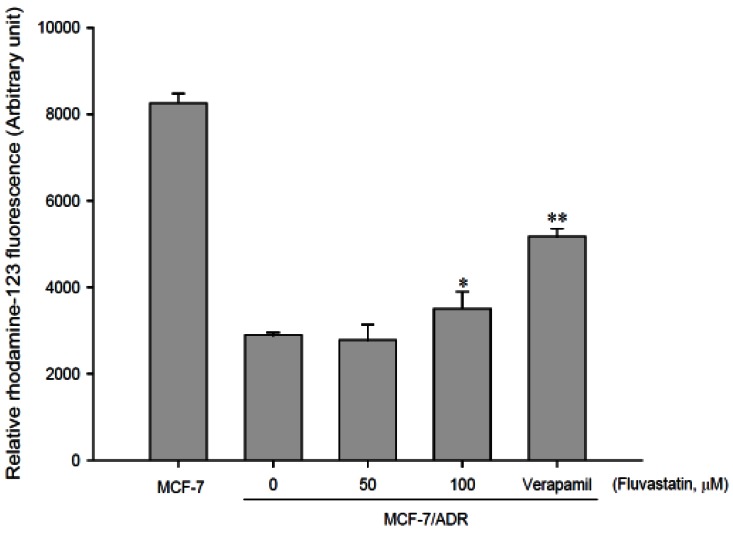
As shown in Fig. 2, accumulation of rhodamine-123, a P-gp substrate, was reduced in MCF-7/ADR cells overexpressing P-gp compared to that in MCF-7 cells lacking P-gp. The concurrent use of fluvastatin enhanced the cellular uptake of rhodamine-123 in a concentration-dependent manner and the uptake was significantly (p<0.01) increased at 100 µM fluvastatin. This result suggests that fluvastatin significantly inhibits P-gp activity.
Determination of blood glucose concentrations
Repaglinide pharmacodynamics were characterized by determining blood glucose levels. Mean blood glucose-time profiles in rats after oral (0.5 mg/kg) and intravenous (0.2 mg/kg) administration of repaglinide in the presence or absence of fluvastatin (1 and 3 mg/kg; n=6, each) are shown in Fig. 3 and 4. Compared to the control group, fluvastatin significantly decreased the blood glucose levels after oral administration of repaglinide and exhibited a significant hypoglycemic effect (Fig. 3); however, there was no significant difference after intravenous administration of re paglinide (Fig. 4).
 | Fig. 3Mean blood glucose-time profiles in rats after oral (0.5 mg/kg) administration of repaglinide in the presence or absence of fluvastatin (1 and 3 mg/kg; n=6, each). Bars represent the standard deviation; Oral administration of 0.5 mg/kg repaglinide (control; solid circles ●), with 1 mg/kg fluvastatin (open circles ○) and with 3 mg/kg fluvastatin (solid triangles ▼) *p<0.05 significant difference compared to control.
|
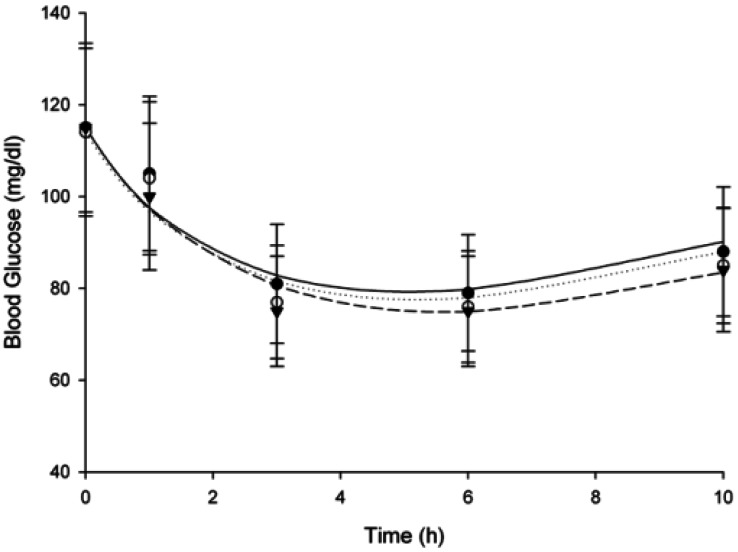 | Fig. 4Mean blood glucose-time profiles in rats after intravenous (0.2 mg/kg) administration of repaglinide in the presence or absence of fluvastatin (1 and 3 mg/kg; n=6, each). Bars represent the standard deviation; Intravenous administration of 0.2 mg/kg repaglinide (control; solid circles ●), with 1 mg/kg fluvastatin (open circles ○) and with 3 mg/kg fluvastatin (solid triangles ▼).
|
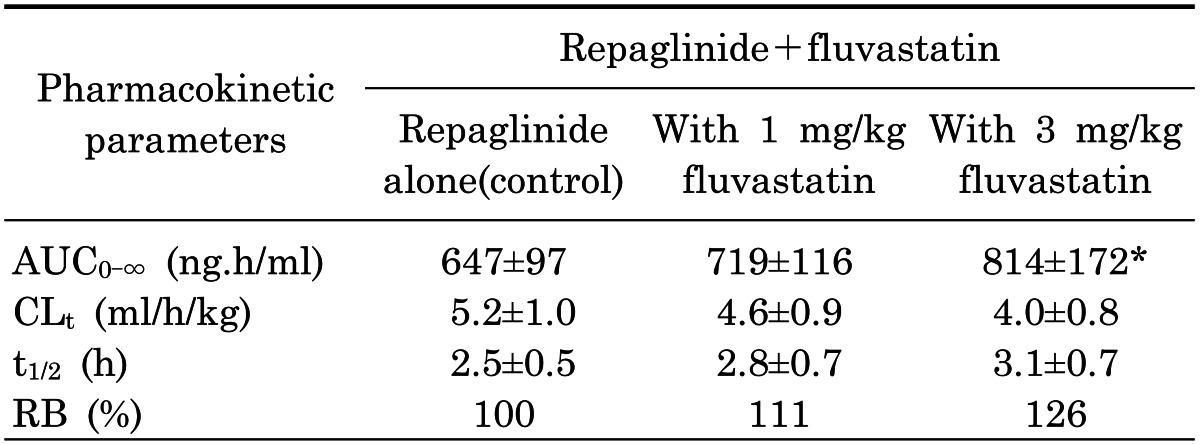
Effect of fluvastatin on the pharmacokinetics of oral repaglinide
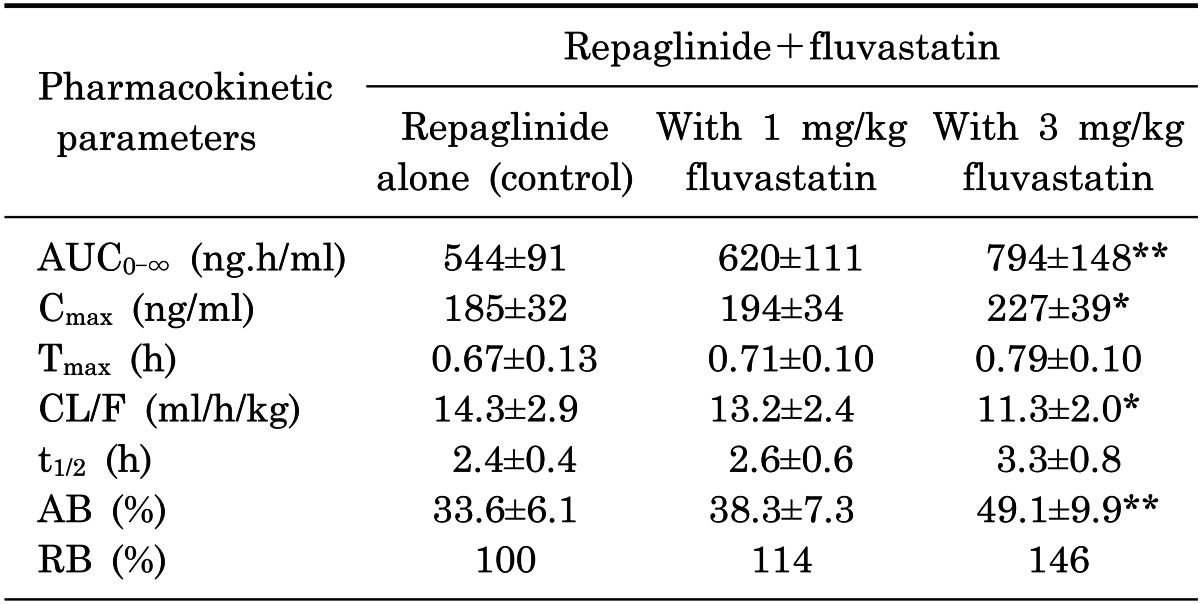
Mean arterial plasma concentration-time profiles of repaglinide following oral administration of repaglinide (0.5 mg/kg) to rats in the presence or absence of fluvastatin (1 and 3 mg/kg) are shown in Fig. 5; the corresponding pharmacokinetic parameters are shown in Table 1. Fluvastatin significantly altered the pharmacokinetic parameters of repaglinide. Compared to the control group (given oral repaglinide alone), fluvastatin significantly increased the area under the plasma concentration-time curve (AUC0-∞; p<0.01 at 3 mg/kg) and the peak plasma concentration (Cmax; p<0.05 at 3 mg/kg) of repaglinide by 45.9% and 22.7%, respectively. Fluvastatin significantly decreased the total body clearance (CL/F) of repaglinide by 21.0% compared to the oral control group.
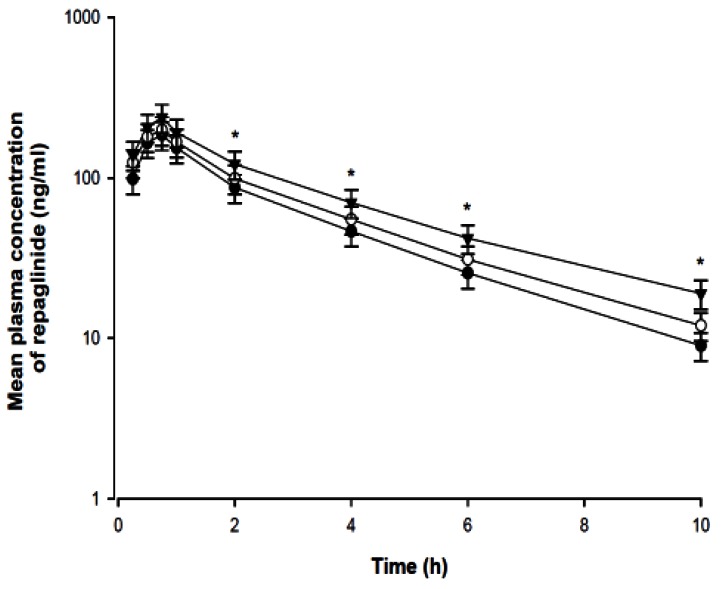 | Fig. 5Mean plasma concentration-time profiles of repaglinide after oral (0.5 mg/kg) administration of repaglinide to rats in the presence or absence of fluvastatin (1 and 3 mg/kg; n=6, each). Bars represent the standard deviation; Oral administration of 0.5 mg/kg repaglinide (control; solid circles ●), with 1 mg/kg fluvastatin (open circles ○) and with 3 mg/kg fluvastatin (solid triangles ▼) *p<0.05 significant difference compared to control.
|
Table 1
Mean (±SD) pharmacokinetic parameters of repaglinide after the oral administration of repaglinide (0.5 mg/kg) to rats in the presence or absence of fluvastatin (1 and 3 mg/kg n=6, each)
Values are mean±SD (n=6); *p<0.05, **p<0.01. Significantly different-compared to control group. AUC0-∞, area under the plasma concentration-time curve from 0 h to infinity; Cmax, peak plasma concentration; Tmax, time to reach peak plasma concentration; CL/F, total body clearance; t1/2, terminal half-life; AB, absolute bioavailability; RB, relative bioavailability.
![]()
Fluvastatin also increased the absolute bioavailability (AB) of repaglinide by 46.1% (3 mg/kg; p<0.01) compared to the oral control group, and the relative bioavailability (RB) of repaglinide by 1.14- to 1.46-fold. There were no significant differences in the time to reach peak plasma concentration (Tmax) and the terminal half-life (t1/2) of repaglinide in the presence of fluvastatin.
Effect of fluvastatin on the pharmacokinetics of intravenous repaglinide
Mean plasma concentration-time profiles of repaglinide after the intravenous administration of repaglinide (0.2 mg/kg) to rats in the presence or absence of fluvastatin (1 and 3 mg/kg) are shown in Fig. 6, while the correlated pharmacokinetic parameters are shown in Table 2. Compared to the control group, fluvastatin (3 mg/kg) significantly (p<0.05) increased the AUC0-∞ (25.8%) of repaglinide. The CLt values of repaglinide tended to decrease, but this trend was not statistically significant. The t1/2 of repaglinide was also increased, but this increase was not significant.
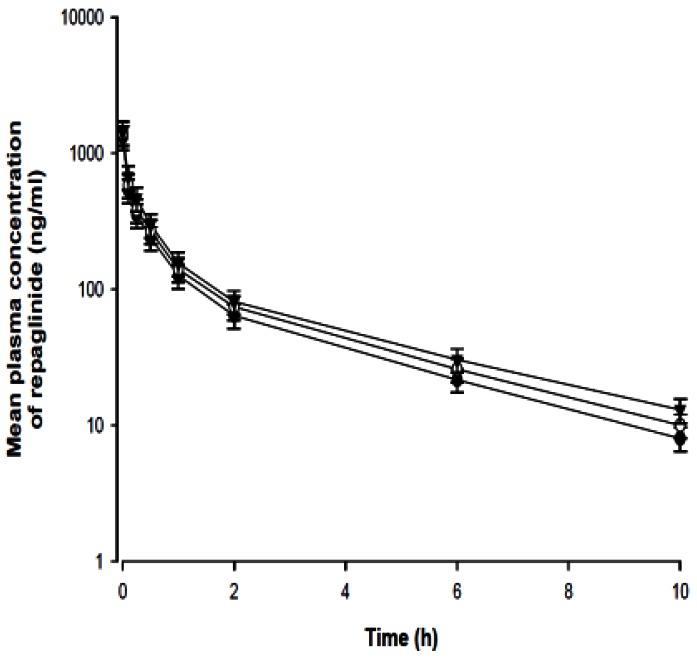 | Fig. 6Mean plasma concentration-time profiles of repaglinide after intravenous (0.2 mg/kg) administration of repaglinide to rats in the presence or absence of fluvastatin (1 and 3 mg/kg; n=6, each). Bars represent the standard deviation; Intravenous administration of 0.2 mg/kg repaglinide (control; solid circles ●), with 1 mg/kg fluvastatin (open circles ○) and with 3 mg/kg fluvastatin (solid triangles ▼).
|
Go to :
DISCUSSION
The prescription of more than one drug as a combination therapy is increasingly common in current medical practice. Cholesterol-lowering agents such as HMG-CoA rductase inhibitors could be co-administered with antidiabetic agents for the treatment of hyperlipidemia induced by diabetes [9,17,21]. Based on the broad overlap in the substrate specificities as well as their co-localization in the small intestine, the primary site of absorption for orally administered drugs, cytochrome P450 (CYP) 3A4 and P-glycoprotein (P-gp) are recognized as a concerted barrier to drug absorption [22,23].
Considering that the drugs used in combination therapy often share the same metabolic pathways or cellular transport pathways, there exists a high potential for pharmacokinetic drug interactions between HMG-CoA reductase inhibitors and antidiabetic agents [9,17].
P-gp is expressed with CYP3A4 and glutathione-S-transferase [24,25], which may have synergistic functions in regulating the bioavailability of many orally ingested compounds. In the small intestine, P-gp is co-localized with CYP3A4 at the apical membrane of the cells [26]. P-gp and CYP3A4 may act synergistically to limit oral absorption and first-pass metabolism [27]. Poor solubility and first-pass metabolism in the liver and epithelial cells of the small intestine result in low bioavailability of repaglinide (56%) [1,2].Considering that repaglinide is a substrate of both CYP3A4 and P-gp [5,6,8,9,28], CYP3A4 and P-gp inhibitors might alter the bioavailability and pharmacokinetics of repaglinide. The inhibitory effect of fluvastatin against CYP3A4-mediated metabolism was confirmed through the use of recombinant CYP3A4 enzyme.
As shown in Fig. 1, fluvastatin inhibited CYP3A4 activity with an IC50 value of 4.1 µM. Furthermore, the cell-based assay using rhodamine-123 indicated that fluvastatin (100 µM) significantly (p<0.05) inhibited P-gp-mediated drug efflux (Fig. 2). CYP3A4 expressed in rat corresponds to and has a similar to the function of CYP3A4 in human [5,29-33]. Therefore, fluvastatin might increa se absorption of repaglinide in the intestine through the inhibition of P-gp and CYP3A4. This paper was only effects of fluvastatin on the pharmacokinetics of repaglinide and it is not drug interaction, so we measured only plasma concentration of repaglinide.
Fluvastatin significantly decreased the blood glucose levels compared to the oral control group. As shown in Table 1, compared to the control group (given oral repaglinide alone), fluvastatin significantly increased the area under the plasma concentration-time curve (AUC0-∞; p<0.01 at 3 mg/kg) and the peak plasma concentration (Cmax; p<0.05 at 3 mg/kg) of repaglinide by 45.9% and 22.7%, respectively.
Fluvastatin significantly increased the absolute bioavailability (AB) of repaglinide by 46.1% and significantly decreased the total body clearance (CL/F) of repaglinide by 21.0% compared to the oral control group. Because fluvastatin considerably increased Cmax and t1/2 of repaglinide, it seems that fluvastatin inhibited the CYP3A4-mediated biotransformation of repaglinide mainly during first-pass metabolism. CYP3A4 is present in considerable quantities in the small-intestine mucosa [34,35], and intestinal CYP3A4 has been shown to play a major role in drug interactions with CYP3A4 inhibitors. These results are consistent with a report by Choi et al. showing that fluvastatin significantly increased the AUC0-∞ and Cmax of oral diltiazem [16].
Our results are consistent with a report by Niemi et al. showing that clarithromycin, a CYP3A4 inhibitor, significantly increased the AUC0-∞ and Cmax of repaglinide and enhanced the repaglinide blood glucose-lowering effect [28], and Kajosaari et al. reported that gemfibrozil and atovastatin significantly increased the AUC0-∞ of repaglinide and enhanced the blood glucose-lowering effect of repaglinide [9], and Kajosaari et al. also reported that co-administration of repaglinide with the CYP3A4 and OATP inhibitor, cyclosporine A significantly increased the plasma concentration of repaglinide in human [6], similarly, fonidipine, a dual inhibitor of CYP3A4 and P-gp, significantly increased the AUC0-∞ and Cmax of oral repaglinide in rats [5]. After intravenous administration of repaglinide with fluvastatin, the AUC0-∞ of repaglinide was significantly increased (p<0.05) by fluvastatin. The CLt values of repaglinide tend to decrease, but this trend was not statistically significant. These results are consistent with a report by Li et al. showing that efonidipine, a dual inhibitor of CYP3A4 and P-gp, significantly increased the AUC0-∞ values of i.v. repaglinide in rats [5].
In summary, fluvastatin enhanced the bioavailability of oral repaglinide in this study. The enhanced bioavailability of repaglinide might be mainly due to rather inhibition of CYP3A4-mediated metabolism of repaglinide in the small intestine and/or liver and to than inhibition of the P-gp efflux transporter in the small intestine and/or to the reduction of total body clearance of repaglinide by fluvastatin. Since the present study indicates the awareness of potential drug interactions in the concomitant use of fluvastatin with repaglinide, the clinical significance of this finding needs to be further evaluated in clinical studies.
Go to :
 XML Download
XML Download