

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Abnormal vascular smooth muscle cell (VSMC) proliferation and migration play important roles in the development and progression of proliferative cardiovascular diseases, including restenosis and atherosclerosis [1,2]. Thus, determination of the mechanisms by which growth factors control cell proliferation is critical for the discovery of compounds capable of intervening in the abnormal proliferation of VSMCs. Receptor tyrosine kinases, including platelet-derived growth factor (PDGF) receptor (PDGF-R), play important roles in VSMC proliferation [3], and so inhibition of abnormal hyperactive PDGF-R-mediated signaling pathways is a major target for the management of abnormal VMSC proliferation.
The binding of PDGF produced by activated macrophages, VSMCs, and endothelial cells with PDGF-R leads to receptor autophosphorylation, subsequently activating phosphatidylinositol 3-kinase (PI3K), phospholipase Cγ (PLCγ), and extracellular regulated kinases 1/2 (ERK1/2) [4]. Active PI3K is coupled to its downstream target, Akt kinase [5], and this PI3K-Akt pathway is involved in the antiapoptotic activity of PDGF in VSMCs [6]. PLCγ1 is a downstream molecule in the PDGF-dependent signal transduction pathway [7]. ERK1/2 mediates PDGF-stimulated VSMC proliferation [8]. As these three major signaling molecules are involved in PDGF-induced VSMC proliferation, inhibition of signaling pathways at the PDGF-R level may be a good strategy for control of VSMC proliferation.
Naphthoquinone derivatives are known to have antitumor, antiviral, antifungal, antimycobacterial, and antiplatelet activities [9-13]. Although the naphthoquinone analog, 2,3-dimethoxy-1,4-naphthoquinone (DMNQ), is cytotoxic [14-18], a recent report indicated that 2-decylamino-DMNQ has an inhibitory effect on PDGF-induced VSMC proliferation with no cytotoxicity at the concentration tested [19]. This derivative acts via cell cycle arrest at the G0/G1 phase. This observation provides a compelling reason to synthesize new naphthoquinone derivatives with antiproliferative actions at the PDGF-R level. Therefore, the aim of this study was to investigate the ability of a newly synthesized naphthoquinone derivative, 5,8-dimethoxy-2-nonylamino-naphthalene-1,4-dione (2-nonylamino-DMNQ) (Fig. 1A), to inhibit PDGF-stimulated VSMC proliferation with the blockade of PDGF-R. Our results reveal that 2-nonylamino-DMNQ inhibits PDGF-dependent receptor autophosphorylation and downstream signaling pathways. These results suggest that 2-nonylamino-DMNQ may be a candidate agent for the prevention and treatment of abnormal VSMC proliferation.
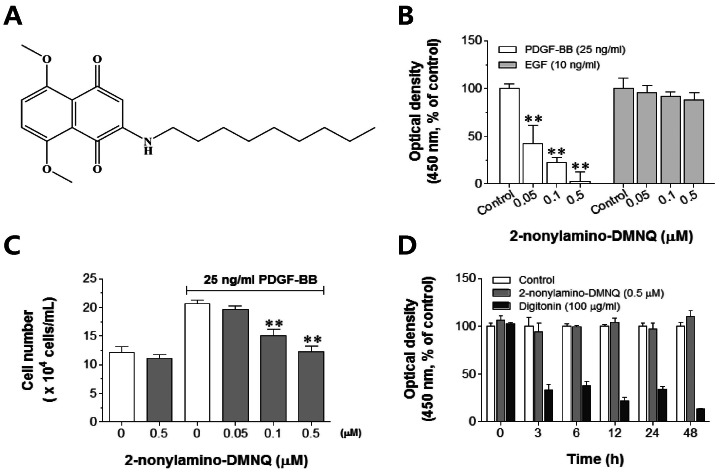 | Fig. 1Effects of 2-nonylamino-DMNQ on VSMC proliferation and viability. (A) The chemical structure of 2-nonylamino-DMNQ. (B) The ability of 2-nonylamino-DMNQ to regulate both PDGF- and EFG-induced VSMC proliferation. VSMCs cultured in serum-free medium were incubated with various concentrations of 2-nonylamino-DMNQ (0.05~0.5 µM) for 24 h and then stimulated with PDGF-BB (25 ng/ml) or EGF (10 ng/ml). The optical density at 450 nm was determined using a microplate reader. (C) The ability of 2-nonylamino-DMNQ to regulate cell counts. VSMCs cultured in serum-free medium were treated with various concentrations of 2-nonylamino-DMNQ for a further 24 h, stimulated with PDGF-BB (25 ng/ml), and counted using a hemocytometer. (D) Effects of 2-nonylamino-DMNQ on cell viability. VSMCs cultured in serum-free medium were incubated with control (DMSO, 1%), or 2-nonylamino-DMNQ (0.5 µM) or digitonin (100 µg/ml) for the indicated periods, and optical densities at 450 nm were measured. Values are expressed as means±S.E.M. of four similar and independent experiments. **p<0.01, statistically significant differences compared to PDGF control (PDGF-BB-stimulated, but no 2-nonylamino-DMNQ) or EGF control (EGF-stimulated, but no 2-nonylamino-DMNQ).
|
Go to : 
METHODS
Materials
Cell culture materials were purchased from Invitrogen (Carlsbad, CA, USA). PDGF-BB and epidermal growth factor (EGF) were obtained from Upstate Biotechnology (Lake Placid, NY, USA). Digitonin and SU6656 were acquired from Sigma Aldrich Inc. (St. Louis, MO, USA). [3H]-Thymidine was purchased from AgenBio, Ltd. (Seoul, Korea). Anti-β-actin, anti-cyclin E, anti-cyclin D, anti-CDK2, anti-CDK4, anti-phospho-retinoblastoma protein (pRb), and anti-phospho-proliferating cell nuclear antigen (PCNA) antibodies were purchased from AbFrontier (Geumcheon, Seoul, Korea). Anti-PDGF-Rβ, anti-phospho-PDGF-Rβ chain (Tyr751, Tyr1021), anti-phospho-STAT3 (Tyr705), anti-ERK1/2, anti-phospho-ERK1/2, anti-Akt, anti-phospho-Akt, anti-PLCγ1, and anti-phospho-PLCγ1 antibodies were obtained from Cell Signaling Technology, Inc. (Beverly, MA, USA). AG1295 was purchased from Enzo Life Sciences, Inc. (Ann Arbor, MI, USA). Anti-phospho-PDGF-Rβ chain (Tyr579, Tyr716) antibodies were obtained from Millipore Corporation (Billerica, MA, USA). Other chemicals were of analytical grade.
Cell proliferation and viability assays
Both direct cell counting and nonradioactive colorimetric WST-1 assay (premix WST-1, Takara, Japan) were used to measure VSMC proliferation, as described previously [19,21,22]. For direct cell counting, VSMCs were treated with various concentrations of 2-nonylamino-DMNQ for 24 h in serum-free medium and stimulated with PDGF-BB (25 ng/ml). 2-Nonylamino-DMNQ was dissolved in dimethylsulfoxide (DMSO), the final concentration of which in medium did not exceed 0.05%. After 24 h, the trypsinized VSMCs were counted using a hemocytometer under a light microscope. For nonradioactive colorimetric WST-1 assays, VSMCs were treated with PDGF-BB (25 ng/ml) or EFG (10 ng/ml), and all experimental procedures were performed as recommended by the respective manufacturers. Results are expressed as percentages of controls (VSMCs treated with either growth factor, but not test compound). Cell viability assays were performed as described previously [19,21,22]. The absorbance at 450 nm of VSMCs treated with 0.5 µM 2-nonylamino-DMNQ or 100 µg/ml digitonin, as a cytotoxic control, for a given time were measured using a microplate reader (Packard Instrument Co., Downers Grove, IL, USA).
[3H]-Thymidine incorporation assay
[3H]-Thymidine incorporation assay was performed to measure DNA synthesis, as described previously [19,21,22]. Under the stimulatory condition with addition of PDGF-BB (25 ng/ml) in serum-free medium, VSMCs were treated with [3H]-thymidine (2 µCi/ml) for 4 h, which was terminated by washing with phosphate-buffered saline (PBS) containing 10% trichloroacetic acid and ethanol/ether (1 : 1, v/v). Acid-insoluble [3H]-thymidine was collected, mixed with 3 ml of scintillation cocktail (Ultimagold; Packard Bioscience, Meriden, CT, USA), and quantified using a liquid scintillation counter (LS3801; Beckman, Düsseldorf, Germany).
Analysis of cell cycle progression
Cell cycle progression was determined as described previously [19,21,22]. VSMCs were treated with PDGF-BB (25 ng/ml) for 24 h, trypsinized, and centrifuged at 1,500×g for 7 min. The pellets were suspended in 1 ml of 1×PBS, washed twice, and fixed with 70% ethanol for 48 h. After centrifuging the fixed cells at 15,000×g for 5 min, the pellets were stained with 500 µl of propidium iodide (PI) solution (50 µg/ml PI in sample buffer containing 100 µg/ml of RNase A), and the fluorescence intensity of incorporated PI reflecting the individual nuclear DNA content was measured with a FACSCalibur flow cytometer (Becton & Dickinson Co., Fullerton, CA, USA). The ratio of G0/G1, S, and G2/M phases of the cell cycle was determined using ModFit LT software (Verity Software House, Topsham, ME, USA).
Western blotting
Western blotting was performed as described previously [19,21,22]. VSMCs were treated with 25 ng/ml PDGF-BB for phosphorylation of PDGF-Rβ (3 min), ERK 1/2 and PLCγ1 (5 min), STAT3 (10 min), AKt (15 min), and pRb (24 h), and for the expression of cyclin D1/E, CDK2/4, and PCNA (24 h), respectively. The detected proteins were normalized relative to β-actin or the respective total proteins. Band intensities were quantified using the Quantity One program (Bio-Rad, Hercules, CA, USA).
Statistical analysis
Data are expressed as means±standard error of the mean (S.E.M.). One-way ANOVA was used for multiple comparisons (GraphPad, San Diego, CA, USA). If a significant variation between treated groups was found, Dunnett's test was applied. In all analyses, p<0.05 was taken to indicate statistical significance.
Go to :
RESULTS
Effects of 2-nonylamino-DMNQ on VSMC proliferation and viability
To examine the effects of 2-nonylamino-DMNQ on VSMC proliferation, a nonradioactive colorimetric WST-1 assay was performed in VSMCs stimulated with PDGF-BB (25 ng/ml) or EGF (10 ng/ml). The data shown in Fig. 1B indicate that 2-nonylamino-DMNQ inhibited PDGF-BB-stimulated proliferation of VSMCs in a concentration-dependent manner. Interestingly, 2-nonylamino-DMNQ did not inhibit EGF-stimulated proliferation of VSMCs. These results suggest that the inhibitory action of 2-nonylamino-DMNQ may be selective for PDGF-induced VSMC proliferation.
Fig. 1C shows that the number of VSMCs was significantly increased after 25 ng/ml PDGF-BB treatment (20.6±1.9×104 cells/well) compared to the non-stimulated control (12.1±0.5×104 cells/well). As expected, the inhibitory action of 2-nonylamino-DMNQ in PDGF-treated VSMCs was concentration-dependent. As the concentration of 2-nonylamino-DMNQ was increased to 0.05, 0.1, and 0.5 µM, cell numbers were significantly reduced to 19.5±1.3, 15.0±1.1, and 12.2±1.0×104 cells/well, respectively. These results indicate that 2-nonylamino-DMNQ inhibits PDGF-stimulated VSMC proliferation.
To investigate the negative influence of 2-nonylamino-DMNQ on cell viability, VSMCs were incubated with the highest concentration of this compound for various times up to 48 h. As shown in Fig. 1D, 0.5 µM 2-nonylamino-DMNQ did not exert any cytotoxicity irrespective of incubation time. Digitonin (100 µg/ml) was used as a positive cytotoxic control. These results indicate that the antiproliferative action of 2-nonylamino-DMNQ in VSMCs was not attributable to cytotoxicity.
Effects of 2-nonylamino-DMNQ on DNA synthesis and cell cycle progression
To determine the effects of 2-nonylamino-DMNQ on DNA synthesis, [3H]-thymidine incorporation was measured in PDGF-treated VSMCs. The data shown in Fig. 2A indicate that 2-nonylamino-DMNQ inhibited [3H]-thymidine incorporation in a dose-dependent manner. PDGF (25 ng/ml) stimulated [3H]-thymidine cpm/well values by 192,885.5±2,886.5/well compared to the control (non-PDGF-treated, 1,322.3±445.6/well). The inhibition rates of 0.05, 0.1, and 0.5 µM 2-nonylamino-DMNQ were 25.92±8.79%, 50.73±9.17% and 60.85±5.23%, respectively. These results indicate that 2-nonylamino-DMNQ inhibits DNA synthesis in PDGF-stimulated VSMCs.
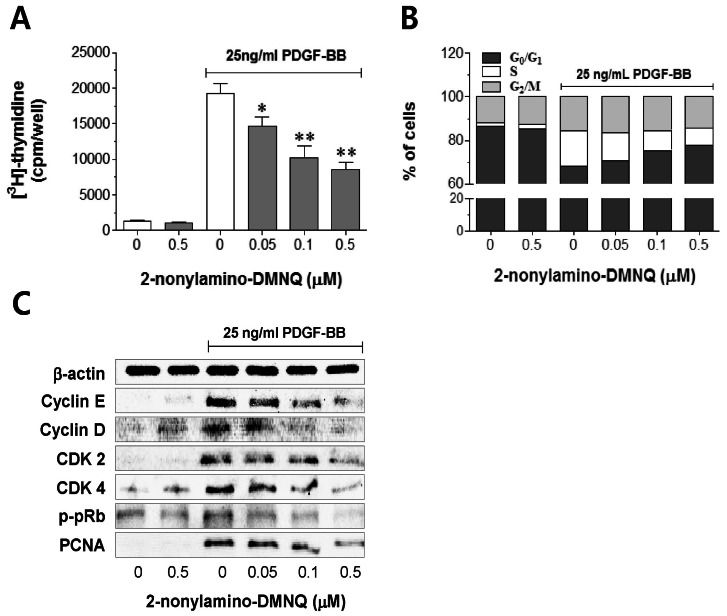 | Fig. 2Effects of 2-nonylamino-DMNQ on [3H]-thymidine incorporation, cell cycle progression, and cell cycle regulatory proteins in PDGF-stimulated VSMCs. (A) The ability of 2-nonylamino-DMNQ to regulate [3H]-thymidine uptake. PDGF-BB (25 ng/ml)-stimulated VSMCs were treated with or without the indicated concentrations of 2-nonylamino-DMNQ for 24 h, and then incubated with [3H]-thymidine (2 µCi/ml) for 4 h. Radioactivity was determined using a liquid scintillation counter. Data are representative of four independent experiments, and are expressed as means±S.E.M. *p<0.05, **p<0.01, statistically significant differences compared to PDGF control (PDGF-BB-stimulated, but no 2-nonylamino-DMNQ). (B) The ability of 2-nonylamino-DMNQ to regulate cell cycle progression. VSMCs in serum-free medium were incubated with or without 2-nonylamino-DMNQ (0.05~0.5 µM) for 24 h, stimulated with 25 ng/ml PDGF-BB for 24 h, harvested, and subjected to flow cytometric analysis of DNA content as described in Methods. Each point was derived from a representative experiment, in which data from at least 11,000 events were obtained. The results are representative of three similar and independent experiments. (C) The ability of 2-nonylamino-DMNQ to regulate cell cycle regulatory proteins. Serum-starved VSMCs were pretreated with or without the indicated concentrations of 2-nonylamino-DMNQ for 24 h, stimulated with PDGF-BB (25 ng/ml) for a further 24 h, and the expression of cyclin E, cyclin D CDK2, CDK4, and PCNA, and the phosphorylation of pRb were measured as described in Methods. Total β-actin was used for normalization. Images are representative blots of four similar and independent experiments.
|
To investigate the mechanism of the anti-proliferative action of 2-nonylamino-DMNQ, a cell cycle progression study was carried out in PDGF-stimulated VSMCs. As shown in Fig. 2B, 2-nonylamino-DMNQ dose-dependently increased the proportion of cells in G0/G1 phase. VSMC serum deprivation resulted in an approximately 87.0±1.9% synchronization of the cell cycle in the G0/G1 phase, and addition of PDGF decreased the G0/G1 phase and the percentage of cells in the S phase increased from 2.0±1.2% to 16.2±2.7%. This cell cycle progression was significantly blocked by 2-nonylamino-DMNQ; the percentage of cells in the S phase was reduced by 13.2±1.8%, 9.3±1.7% and 8.1±0.5% at concentrations of 0.05, 0.1, and 0.5 µM, respectively. This observation suggests that 2-nonylamino-DMNQ may act in the early events of the cell cycle to affect DNA synthesis induced by PDGF.
To explore the mechanism by which 2-nonylamino-DMNQ induces cell cycle arrest, cyclin D, cyclin E, CDK 4, and CDK 2 expression were determined. The data shown in Fig. 2C indicate that 2-nonylamino-DMNQ significantly suppressed the expression of cyclin E, cyclin D, CDK 2, and CDK 4 in a concentration-dependent manner. In addition, 2-nonylamino-DMNQ inhibited the hyperphosphorylation of pRb and the expression of PCNA in a concentration-dependent manner. These observations suggest that 2-nonylamino-DMNQ inhibits the cell cycle in S phase by arresting cells in the G0/G1 phase.
Effects of 2-nonylamino-DMNQ on the activation of PDGF receptor tyrosine kinase
To investigate the molecular mechanism underlying the inhibitory action of 2-nonylamino-DMNQ in PDGF-induced proliferation, the phosphorylation of PDGF-Rβ (Tyr751) was measured in PDGF-stimulated VSMCs. As shown in Fig. 3A, PDGF-Rβ was significantly phosphorylated by the addition of PDGF, and this was concentration-dependently inhibited by 2-nonylamino-DMNQ in PDGF-stimulated VSMCs. The inhibition of PDGF-induced PDGF-Rβ phosphorylation by the highest concentration (0.5 µM) of 2-nonylamino-DMNQ was comparable to the addition of 5 µM AG1295, a selective inhibitor of PDGF receptor tyrosine kinase (Fig. 3B) [23].
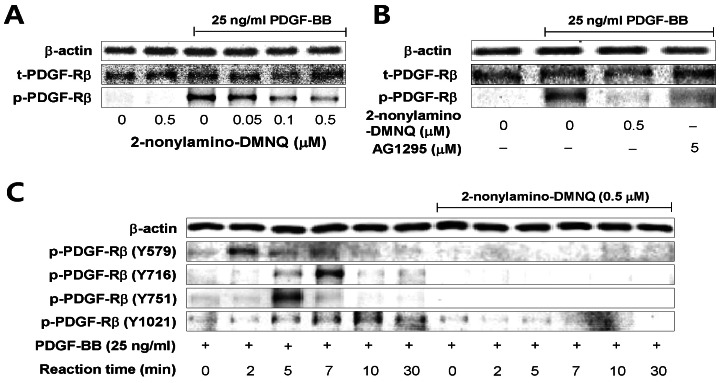 | Fig. 3Effects of 2-nonylamino-DMNQ on PDGF-induced PDGF-Rβ activation in VSMCs. (A) The ability of 2-nonylamino-DMNQ to inhibit PDGF-Rβ activation induced by PDGF. Serum-starved VSMCs were treated with or without the indicated concentrations of 2-nonylamino-DMNQ for 24 h, stimulated with PDGF-BB (25 ng/ml) for 3 min, and the phosphorylation of PDGF-Rβ was measured as described in Methods. (B) Comparison of the ability of 2-nonylamino-DMNQ and AG1295 to inhibit PDGF-induced PDGF-Rβ activation. VSMCs cultured in serum-free medium were incubated with or without the indicated concentrations of 2-nonylamino-DMNQ or AG1295, a selective inhibitor of PDGF receptor kinase, for 24 h, stimulated with PDGF-BB (25 ng/ml) for 3 min, and the phosphorylation of PDGF-Rβ was measured as described in Methods. (C) The ability of 2-nonylamino-DMNQ to regulate phosphorylation at four different tyrosine residues in PDGF-stimulated PDGF-Rβ. Serum-starved VSMCs were incubated with or without 0.5 µM 2-nonylamino-DMNQ for 24 h. After reacting VSMCs with PDGF-BB (25 ng/ml) for the indicated periods, the phosphorylation of PDGF-Rβ at Tyr579, Tyr716, Tyr751, and Tyr1021 residues was measured as described in Methods. Images are representative blots of three similar and independent experiments.
|
To further examine the effects of 2-nonylamino-DMNQ on phosphorylation of four different tyrosine residues in PDGF-Rβ, tyrosine position-specific anti-phospho-PDGF-Rβ antibodies were applied to PDGF-stimulated VSMCs in the absence or presence of 2-nonylamino-DMNQ (0.5 µM) for times up to 30 min. The data shown in Fig. 3C present that 2-nonylamino-DMNQ completely blocked the phosphorylation of four tyrosine residues (Tyr579, Tyr716, Tyr751, Tyr1021) in PDGF-Rβ. The highest level of Tyr579 phosphorylation in PDGF-Rβ occurred at 2 min, Tyr716 at 7 min, Tyr751 at 5 min, and Tyr1021 at 10 min. These results indicate that 2-nonylamino-DMNQ inhibited the phosphorylation of PDGF-Rβ tyrosine kinase, and subsequently suppressed VSMC proliferation.
Effects of 2-nonylamino-DMNQ on the phosphorylation of STAT3, ERK1/2, Akt, and PLCγ
To examine the influence of 2-nonylamino-DMNQ on the PDGF-Rβ-mediated downstream signaling pathway, the phosphorylation of STAT3, ERK1/2, Akt, and PLCγ1 was determined in PDGF-stimulated VSMCs. The data shown in Fig. 4A indicate that 2-nonylamino-DMNQ concentration-dependently inhibited the STAT3 phosphorylation (Tyr705) induced by PDGF. The inhibitory action induced by the highest concentration of 2-nonylamino-DMNQ (0.5 µM) was comparable to pretreatment with 2 µM SU6656, a Src family kinase inhibitor [24]. This observation indicates that 2-nonylamino-DMNQ inhibits PDGF-Rβ-mediated Src-dependent STAT3 activation in PDGF-stimulated VSMCs.
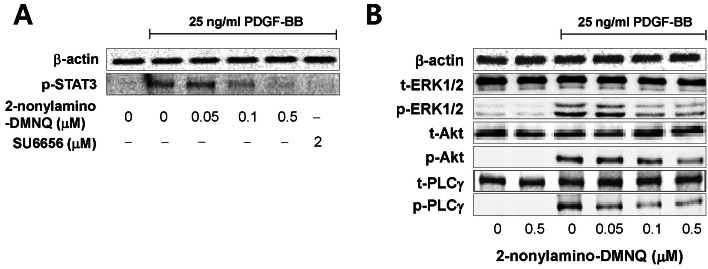 | Fig. 4Effects of 2-nonylamino-DMNQ on STAT3, ERK1/2, Akt and PLCγ1 phosphorylation in PDGF-stimulated VSMCs. (A) The ability of 2-nonylamino-DMNQ to regulate STAT3 activation. VSMCs cultured in serum-free medium were incubated with or without the indicated concentrations of 2-nonylamino-DMNQ or SU6656, a Src family kinase inhibitor, for 24 h, stimulated with 25 ng/ml PDGF-BB for 10 min, and the phosphorylation of STAT3 was measured as described in Methods. Images are representative blots of four similar and independent experiments. (B) The ability of 2-nonylamino-DMNQ to regulate ERK1/2, Akt, and PLCγ1 activation. Serum-starved VSMCs were treated with or without the indicated concentrations of 2-nonylamino-DMNQ for 24 h, stimulated with 25 ng/ml PDGF-BB for 5 min (ERK1/2 and PLCγ1) or 15 min (Akt), and the ERK1/2, Akt and PLCγ1 phosphorylation were measured as described in Methods. Images are representative blots of four similar and independent experiments.
|
The data shown in Fig. 4B present that 2-nonylamino-DMNQ concentration-dependently inhibited the phosphorylation of ERK1/2, Akt, and PLCγ1 in PDGF-stimulated VSMCs. Total amounts of ERK1/2, Akt, and PLCγ1 were unaffected by 2-nonylamino-DMNQ. These results indicate that 2-nonylamino-DMNQ inhibits PDGF-Rβ-mediated ERK1/2, Akt, and PLCγ1 activation induced by PDGF treatment.
Go to :
DISCUSSION
The major finding of this study is that 2-nonylamino-DMNQ inhibits PDGF-induced VSMC proliferation by blocking the autophosphorylation of PDGF-Rβ and subsequently suppressing PDGF-Rβ-mediated signaling pathways. This is the first report linking the antiproliferative action of a novel DMNQ-derivative, 2-nonylamino-DMNQ, with the underlying mechanism at the receptor level in PDGF-stimulated VSMCs. Thus, our results suggest that 2-nonylamino-DMNQ may be an effective tool to control PDGF-Rβ activity and the PDGF-Rβ-mediated signaling transduction pathway for the management of abnormal VSMC proliferation in atherosclerosis and vascular restenosis after angioplasty.
Cellular proliferation is regulated primarily by the cell cycle. The G1 phase of the cell cycle is a major point of control for cell proliferation in mammalian cells [25] and the cyclin E/CDK 2 and cyclin D/CDK 4 complexes are important for the G1-S phase transition [26]. These complexes subsequently induce the phosphorylation of pRb, followed by DNA synthesis [27]. PCNA is a phosphor-pRb-mediated gene product that is synthesized in the early G0/G1 and S phases of the cell cycle [28]. Our results demonstrated that 2-nonylamino-DMNQ suppresses [3H]-thymidine incorporation (Fig. 2A), and that this inhibition of DNA synthesis is due to the inhibition of cyclin E, cyclin D, CDK2, CDK4, and PCNA expression, and the phosphorylation of pRb (Fig. 2C). Indeed, 2-nonylamino-DMNQ increases the proportion of cells in the G0/G1 phase (Fig. 2B). These data indicated that 2-nonylamino-DMNQ inhibits VSMC proliferation by suppressing cell cycle arrest at the G0/G1 phase.
EGF was shown to induce VSMC proliferation by stimulating Rac1 and p21-activated kinase [29]. However, 2-nonylamino-DMNQ has no action in EGF-stimulated VSMCs (Fig. 1B), indicating that its inhibitory action is PDGF-selective. The PDGF-Rβ phosphorylation induced by PDGF was inhibited by 2-nonylamino-DMNQ (Fig. 3A), and this inhibitory action was similar to that of a selective inhibitor of PDGF-R tyrosine kinase, AG1295 (Fig. 3B), indicating that the inhibitory action of 2-nonylamino-DMNQ does occur at the receptor level. Importantly, the mechanism underlying this activity is specific blockade of the phosphorylation of tyrosine residues (Tyr579, Tyr716, Tyr751, and Tyr1021) in the PDGF-Rβ chain (Fig. 3C). The phosphotyrosine residue at Tyr579 is known to directly mediate the interaction with Src family kinases [30], which subsequently activate STAT3-mediated Myc expression for PDGF-induced mitogenesis [31]. Phosphorylated Tyr716 is important for binding of growth factor receptor-bound protein 2 (GRB2) [32], which induces the translocation of Sos to the plasma membrane, and GRB2/Sos stimulates the Ras/MAPK (ERK1/2) pathway [33]. Tyr751 in activated PDGF-R is known to be a docking site for the PI3K/Akt pathway [34], and PLC signaling is dependent on the autophosphorylation of PDGF-R at Tyr1021 [35]. Thus, 2-nonylamino-DMNQ directly inhibits the autophosphorylation of PDGF-R at four different tyrosine residues and leads to the regulation of downstream signaling pathways, including STAT3, ERK1/2, PI3K/Akt, and PLCγ, which is an important mechanism for the antiproliferative action of 2-nonylamino-DMNQ in PDGF-stimulated VSMCs.
The DMNQ toxic effects previously reported were mainly in liver and lung by inducing oxidative stress. DMNQ caused an extensive glutathione (GSH) depletion accompanied by glutathione disulfide (GSSG) formation, preceding loss of viability in isolated hepatocytes [14], and moreover, decreased in intracellular adenosine 5'-triphosphate (ATP) in γ-glutamyl transpeptidase (GGT)-overexpressing stable cell line, causing continuously generate H2O2 and oxidative injury [15]. Another quinonoid drug, menadione (2-methyl-1,4-naphthoquinone), also showed cytotoxicity in isolated hepatocytes [16]. Similarly, in lung epithelial cells, DMNQ increased GGT mRNA content accompanied by increased GGT specific activity [17], and γ-glutamylcysteine synthetase (γ-GCS) activity [18]. Although this study did not cover these cytotoxicities in both organs, our results indicated that the highest concentration of 2-nonylamino-DMNQ showed the maximal antiproliferative action in PDGF-stimulated VSMCs did not exert any cytotoxicity at least within 48 h incubation (Fig. 1D). This indicates that 2-nonylamino-DMNQ has an antiproliferative property in PDGF-stimulated VSMCs without any toxicity.
In summary, our data indicated that a novel naphthoquinone derivative, 2-nonylamino-DMNQ, inhibits PDGF-stimulated VSMC proliferation without any cytotoxicity, and induces cell cycle arrest at the G0/G1 phase by suppressing the expression of cyclin D, cyclin E, CDK4, CDK2, and PCNA, and the phosphorylation of pRb. The mechanism underlying this antiproliferative action is the inhibition of autophosphorylation of PDGF-Rβ and subsequent downregulation of PDGF-Rβ-mediated signaling pathways, including STAT3, ERK1/2, Akt, and PLCγ. Thus, 2-nonylamino-DMNQ may be useful to control abnormal proliferation of VSMCs at the receptor level for the management of vascular hyperproliferative disorders.
Go to :
 XML Download
XML Download