

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Alzheimer's disease (AD) is the most prevalent neurodegenerative disease worldwide. Its pathological features include synaptic degeneration, senile plaques and neurofibrillary tangles, which eventually lead to neuronal loss [1]. The major components of senile plaque are amyloid-β peptide (Aβ), which are derived by proteolytic cleavage of amyloid precursor protein (APP), a type I membrane spanning protein. Two enzymes, beta-site APP-cleaving enzyme 1 (BACE1; originally referred to as BACE) and BACE2, are involved in Aβ production. BACE1 is mainly responsible for β-cleavage of APP in vivo, and overexpression of BACE1 increases the production of Aβ in vitro. Therefore, BACE1 is considered the pivotal component in Aβ production [2]. Since less than 10% of total AD cases are associated with genetic factors [3], it is extremely important to investigate how environmental factors contribute to the pathogenesis of this disease.
Although the cause or causes of AD are not yet known, most experts agree that AD probably develops as a result of multiple factors rather than a single cause. The greatest risk factor for AD is advancing age [4]. The risk of AD dramatically increases in individuals beyond the age of 70, and it is predicted that the incidence of AD will increase three-fold within the next 50 years [2]. Recent findings suggest that age-related increases in oxidative stress account for aspects of AD [5]. Some evidence demonstrates that oxidative stress is an early event in AD, occurring prior to cytopathology, and therefore may play a key pathogenic role in the disease [6]. It has been reported that iron can accelerate the production of Aβ and is found in high concentrations in the amyloid plaque [7].
The influence of metal ions on AD pathogenesis is becoming evident. Although essential for cell function, increased tissue iron can promote tissue oxidative damage to the brain, which is especially vulnerable [8,9]. In the human brain, iron levels increase with age [10,11], and iron contributes to the development of proteinopathies (abnormal deposits of proteins) associated with several prevalent neurodegenerative diseases such as AD, Parkinson's disease (PD), and Dementia with Lewy Bodies (DLB) [8,12]. Iron levels are abnormally elevated in these age-related neurodegenerative diseases, suggesting that increased iron levels may contribute to their age risk-factor as well as impacting the age at onset especially in males [13]. Although a few studies focused on the effects of metal ions on the transcription of APP, their effects on BACE1, the other pivotal component of Aβ production, are still not well understood.
The potential signal transduction pathway involved in the pathology of AD may be the activation of the MAPKs. MAPKs are classified into different subfamilies, and among them, the best known are the extracellular signal regulated kinase (ERK), the c-Jun N-terminal kinase (JNK) and p38 [14]. The ERK-signal pathway results in phosphorylation of Bad at serine-112 (S1112) and -155 (S155) [15]. Increased Bad phosphorylation reduces its translocation to mitochondria, limits the release of mitochondrial cytochrome c [16] and apoptosis-inducing factor, and increases the resistance to apoptosis. It has been reported that prolonged ERK activation is accompanied by the proapoptotic effect of ERK [17], whereas transient activation of ERK protects cells from death [18].
Bcl-2 families are well-characterized regulators of apoptosis, consisting of three distinct subfamilies. The Bcl-2 subfamily contains antiapoptotic proteins, such as Bcl-2 and Bcl-XL, which reduce the release of cytochrome c [19,20]. The Bax subfamily contains proapoptotic proteins such as Bax and Bak, which induce the release of cytochrome c [21]. Furthermore, the ratio of proapoptotic and antiapoptotic Bcl-2 proteins may be a pivotal cue to release cytochrome c from mitochondria.
Oxidative stress induces BACE1 transcription [22], thereby promoting the production of Aβ [23]. Aβ has iron chelating sites [24], thus protecting the cells from oxidative stress. Therefore, in the present study, we investigated the mRNA levels of BACE1/ADAM10 and altered APP carboxyl-terminal processing under ferrous iron treatment in PC12 cells.
METHODS
PC12 cell culture
The PC12 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM; Gibco, NY, USA) supplemented with 10% fetal bovine serum (FBS; Hyclone Laboratories, Logan, UT, USA), 5 µg/ml plasmocin, 100 U/ml penicillin, and 100 µg/ml. For induction of oxidative condition, PC12 cells were treated with FeCl2 (0.05, 0.1, 0.15, 0.2, 0.25, 0.3, and 0.6 mM) and then incubated at 37℃ with 95% air and 5% CO2 for 30 hr.
Cell survival assay
A Cell Counting Kit-8 (CCK-8; Tokyo, Japan) was used in this study. One hundred microliters of cell suspension without FBS was dispensed into a 96-well plate. FeCl2 (0.05, 0.1, 0.15, 0.2, 0.25, 0.3, and 0.6 mM) was added to the plate wells, respectively. The plates were incubated at 37℃ with 95% air/5% CO2 for 30 hr. Then, 10 µl of CCK-8 solution was added to each well of the plate and incubated for 1 hr. Absorbance was measured with a microplate reader (Molecular Devices, Sunnyvale, CA, USA) at 450 nm.
DAPI staining
PC12 cells were washed extensively with serum-free DMEM and replated onto new 6-well plates. Cells were fixed in 4% paraformaldehyde buffered with 0.1 M phosphate (pH 7.3) for 30 min and then washed with phosphate buffered saline (PBS). The cells were permeabilized with 0.3% Triton X-100 for 20 min, washed with PBS, and then stained with 4,6-diamidino-2-phenylindole (DAPI, Santa Cruz Biotechnology, CA, USA) for 10 min. Cells were observed by fluorescence microscopy (Olympus IX71, Japan) at a peak excitation wavelength of 340 nm.
RNA extraction and quantitative real-time PCR
Total RNA was extracted using Trizol reagent (Invitrogen Co., Carlsbad, CA, USA) according to the manufacturer's instructions, and the concentration of total RNA was determined by measuring the absorbance at 260 nm. A first strand complementary DNA (cDNA) was prepared by subjecting total RNA (1 µg) to reverse transcription using moloney murine leukemia virus (MMLV) reverse transcriptase (iNtRON Bio, Sungnam, Kyunggi, Korea) and random primers (9-mers; TaKaRa Bio Inc, Otsu, Shiga, Japan). Each cDNA template (2 µl) was analyzed in triplicate by addition of 10 µl of 2× SYBR® Premix Ex Taq™ (TaKaRa Bio. Inc., Otsu, Shiga, Japan) along with 10 pmol of each primer. Amplification was conducted using a 7,300 Real-Time PCR System (Applied Biosystems, Foster, CA, USA) and the following parameters: denaturation at 95℃ for 5 minutes; 40 cycles of denaturation at 95℃ for 30 seconds; and annealing and extension at 63℃ for 30 seconds and 72℃ for 45 seconds, respectively. The sequences of the oligonucleotides used for amplification were as follows: ADAM10 forward, 5'-AGCAACATCTGGGGACAAAC-3'; ADAM10 reverse, 5'-TTGCACTGGTCACTGTAGCC-3'; BACE1 forward, 5'-TCGCTGTCTACAGTCATCC-3'; BACE1 reverse, 5'-AACAAACGGACCTTCCACTG-3'; β-actin forward, 5'-AGCCATGTACGTAGCCATCC-3'; β-actin reverse, 5'-TTTGATGTCACGCACGATTT-3'. The relative expression levels of ADAM10 and BACE1 (normalized to that of β-actin) for each sample were determined using RQ software (Applied Biosystems). All real-time PCR experiments were repeated twice.
Western blot analysis
Cells were harvested, washed twice with ice-cold PBS, and then resuspended in 20 mM Tris-HCl buffer (pH 7.4) containing protease inhibitors (0.1 mM phenylmethyl-sul fonyl fluoride, 5 µg/ml aprotinin, 5 µg/ml pepstatin A, and 1 µg/ml chymostatin) and phosphatase inhibitors (5 mM Na3VO4, 5 mM NaF). Whole cell lysate was prepared using 20 strokes of a Dounce homogenizer, followed by centrifugation at 13,000×g for 20 min at 4℃. Protein concentration was determined using the BCA assay (Sigma, St Louis, MO, USA). Proteins (50 µg) were separated by 12% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred onto a polyvinylidene difluoride (PVDF) membrane. The membrane was incubated with antibodies directed against the following proteins, as indicated: phosphorylated (p)-ERK (1:1,000), ERK (1:1,000) (Transduction Laboratories, Lexington, KY, USA); p-Bad (S112, 136, 155) (Cell Signaling Technology, Beverly, MA, USA) (1:1,000); Bad, Bax, and Bcl-2 (Santa Cruz Biotechnology, CA, USA) (1:500); GAPDH (1:1,000) (Assay Designs, MI, USA); anti-APP (Santa Cruz Biotechnology, CA, USA) (1:500). Immunoreactive proteins were visualized by exposure to X-ray film. The protein bands were quantified by image-scanning, and optical density was measured using computer software (NIH Image software 1.52), after the data was corrected by background subtraction and normalized to glyceraldehyde 3-phosphate dehydrogenase (GAPDH), an internal control.
RESULTS
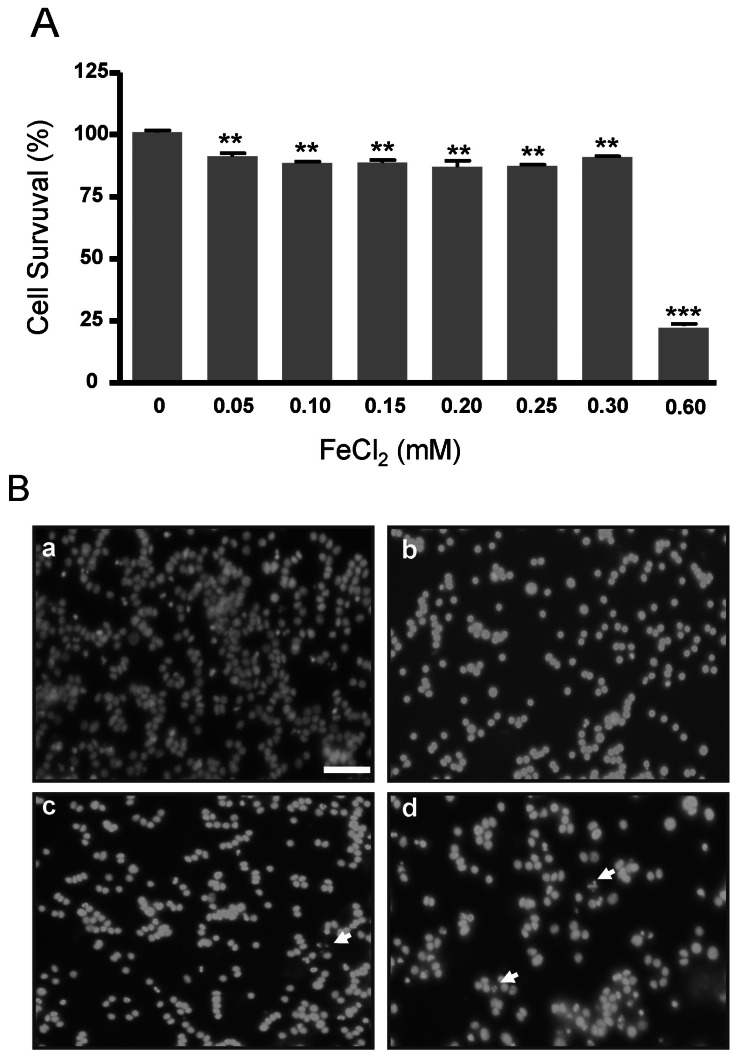
FeCl2 treatment decreases cell survival in PC12 cells
Cell survival and DAPI staining were examined in PC12 cells treated with FeCl2 for 30 h. Cell survival in PC12 cells was significantly decreased in cells treated with 0.05, 0.10, 0.15, 0.2, 0.25, and 0.3 mM FeCl2 (p<0.01), while that in 0.6 mM FeCl2 was significantly reduced by about 75% (p<0.001) (Fig. 1A). This result suggests that the iron concentrations from 0.05 mM to 0.3 mM induced oxidative stress. DAPI staining with enhanced fluorescence was observed in treatment groups of 0.1, 0.2, and 0.3 mM FeCl2, and the nuclei appeared fragmented in 0.2 and 0.3 mM FeCl2 (Fig. 1B).
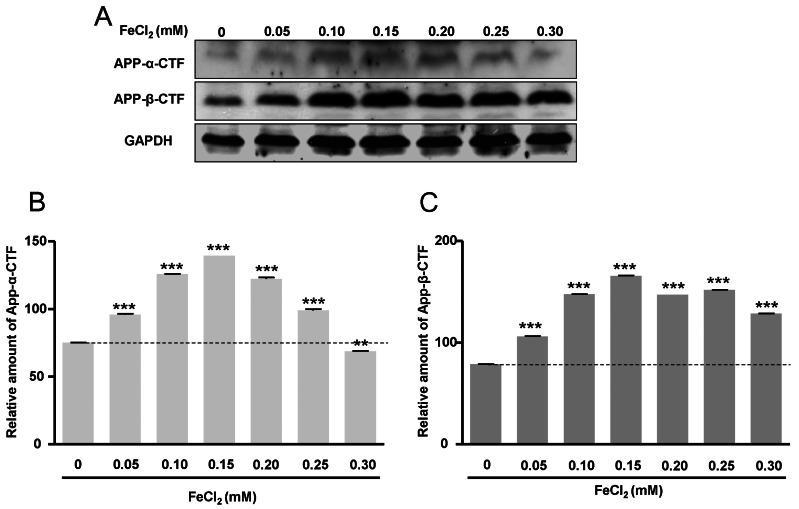
APP-α-CTF and APP-β-CTF expressions with FeCl2 treatment
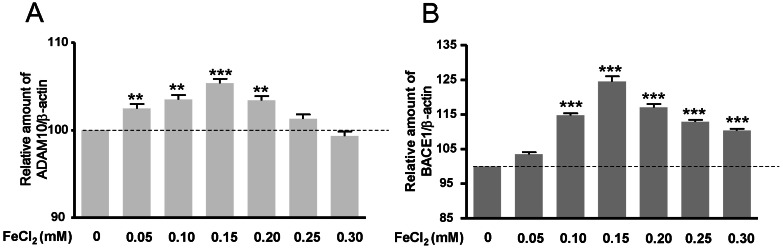
To investigate APP-α-CTF and APP-β-CTF expressions with FeCl2 treatment, Western blot analysis was carried out, and the expression levels of ADAM10 and BACE1 mRNA were determined. Expressions of APP-α-CTF and APP-β-CTF were respectively increased according to FeCl2 concentration (p<0.001) (Fig. 2). In these conditions, the expressions of ADAM10 and BACE1 mRNA were increased significantly relative to the expressions of APP-α-CTF and APP-β-CTF (Fig. 3). This result indicates that increased mRNA of ADAM10 and BACE1 may be associated with the increases of APP-α-CTF and APP-β-CTF expression.
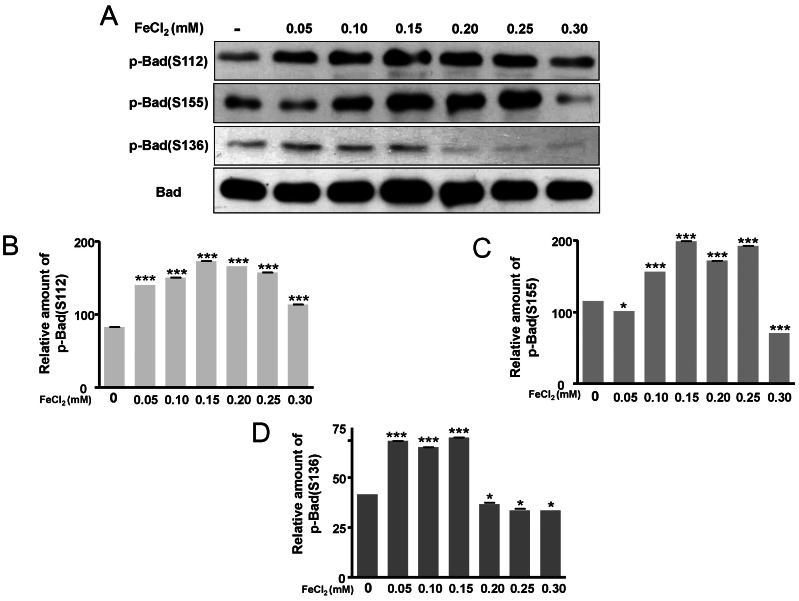
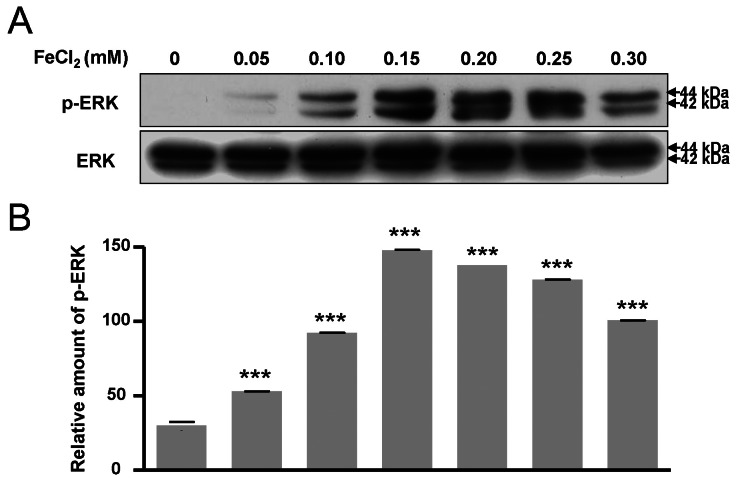
ERK pathway are activated by FeCl2 treatment
The phosphorylation of Bad at two functional sites, S112 and S155, was investigated. The phosphorylation of Bad at S112 and S155 was increased in response to FeCl2 concentration (Fig. 4A~C), and phosphorylation of Bad at S136 was slightly augmented (Fig. 4A, Fig. 4D). This indicates that the translocation of Bad was blocked from cytosol to mitochondria. Thus, mitochondrial cytochrome c release was limited [16]. In this condition, p-ERK was significantly increased (Fig. 5). Therefore, PC12 cells treated with FeCl2 were associated with ERK signal pathway.
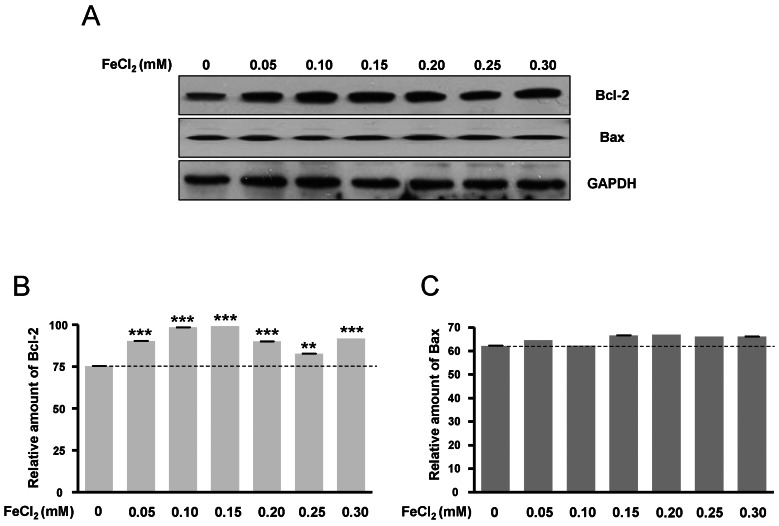
Expressions of Bcl-2 and Bax protein are related to oxidative stress
Bcl-2 protein expression was increased according to FeCl2 concentration (Fig. 6A, B), but Bax protein expression was only slightly changed (Fig. 6A, C). This data was consistent with Fig. 1, suggesting that the expressions of Bcl-2 and Bax protein may be related to oxidative stress.
DISCUSSION
High concentrations of reactive iron increase oxidative-stress induced neuronal vulnerability, and iron accumulation increases the toxicity in the brain [8,9]. Cellular iron mismanagement may cause or exacerbate AD through a variety of cellular mechanisms [8,12]. Iron has been shown to regulate APP synthesis and processing [8,12,13]. In this study, we have shown that iron provokes oxidative stress responses in rat PC12 cells. BACE1 transcription levels increased at the subtoxic concentration of iron that had little effect on cell survival. Transcriptional and translational upregulation of BACE1 can be triggered by oxidative stress [22]. BACE1 knock-out mice fail to produce Aβ [25], indicating that BACE1 is the major enzyme for Aβ production. BACE1 overexpression leads to the production of Aβ in vitro [26]. It has been reported that iron can accelerate the production of Aβ and is found in high concentrations in the amyloid plaque [7]. Therefore, oxidative stress has been considered to have a key pathogenic role in AD [27,28]. However, in the initial stages of AD, Aβ may have neuroprotective effects. Because Aβ possesses iron chelating sites, oxidative stress-induced intracellular iron levels stimulate APP holo-protein expression and the subsequent generation of Aβ as a compensatory response that eventually reduces oxidative stress [29]. This finding indicates that oxidative stress not only causes damage to cellular structures, but also provokes cellular responses. Thus, the physiological pathway of Aβ production may be considered a compensatory or neuroprotective response that reduces oxidative stress damage. These findings also support roles for Aβ in physiological iron regulation [30,31].
We show that ADAM10 is upregulated (under 0.2 mM of FeCl2) and is thought to maintain physiological homeostasis in response to Aβ increase. Production of Aβ is prevented by α-secretase activity which cleaves APP in the Aβ region thereby releasing the soluble N-terminal fragment sAPPα and a C-terminal fragment that is further cleaved by γ-secretase [3]. sAPPα has been shown to exert neurotrophic and neuroprotective effects in vitro [32,33]. However, ADAM10 transcription levels have no remarkable changes when treated with 0.2 mM FeCl2. Apparent discrepancies between our values and those of earlier studies may be due to the effects of Aβ. Aβ may have a decisive role in reducing oxidative stress by chelating ferrous ions. Thus, we suggest that Aβ has a neuroprotective effect on transient oxidative stress or early stage AD.
Our results indicate that ERK activation protects against apoptotic cell death. It has been reported that prolonged ERK activation is accompanied by the proapoptotic effect of ERK [17], whereas a transient activation of ERK protects cells from death [18]. In the present study, we found that activation of the ERK pathway is neuroprotective at subtoxic concentrations. Thus, the protection strategies that target these mechanisms merit further exploration. Furthermore, the kinetics and duration of ERK activation may play an important role in influencing its effect on cell fate.
Bcl-2 plays a protective role against oxidative stress induced by FeCl2 in rat PC12 cells. Bcl-2 is an antiapoptotic protein which reduces cytochrome c release [19]. Bax is a proapoptotic protein which induces cytochrome c release [21]. Overall, cell responses tended to reduce the release of cytochrome c from mitochondria. Thus, the antiapoptotic pathway is predominant. Consistent with this, the ERK pathway is activated. Phosphorylation of Bad at S112 and S155 is upregulated and implies that the antiapoptotic cell response is increased. ERK has been shown to be required for the prevention of apoptosis and the promotion of cell survival through the phosphorylation of proapoptotic Bad [34].
In conclusion, mRNA expressions of ADAM10 and BACE1 were associated with increases of APP-α-CTF and APP-β-CTF expression, respectively, under ferrous iron-induced oxidative stress. This process may involve p-ERK activation.
 XML Download
XML Download