

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
1-EBIO
1-ethyl-2-benzimidazolinone
DAPI
4',6-diamidino-2-phenylindole dihydrochloride
EDHF
endothelium-derived hyperpolarizing factor
eNOS
endothelial NO synthase
FBS
fetal bovine serum
HUVEC
human umbilical vein endothelial cell
Kv
voltage-dependent K+ channel
LPC
lysophosphatidylcholine
NAC
N-acetyl-L-cysteine
pERK
phosphorylated extracellular signal-regulated kinase
ROS
reactive oxygen species
REST
repressor element-1-silencing transcription factor
SOD
superoxide dismutase
TBHP
tert-butyl hydroperoxide
X/XO
xanthine/xanthine oxidase mixture
INTRODUCTION
Vascular endothelial cells are in contact with blood cells and vascular smooth muscle cells, and are therefore constantly exposed to reactive oxygen species (ROS) that are released from these cell types on activation [1,2]. Moreover, endothelial cells themselves generate ROS by stimulation with various substances in plasma [1,2]. ROS play a key role in the physiological and pathological processes in endothelial cells; hydrogen peroxide upregulates endothelial NO synthase (eNOS) [3], and serves as an endothelium-derived hyperpolarizing factor (EDHF) that mediates vascular relaxation [4,5]. Conversely, superoxide impacts endothelial function by downregulating the expression of eNOS [3], thus mediating vascular contraction [6,7]. In addition, excessive ROS production damages endothelial cells, leading to endothelial dysfunction [1].
ROS may regulate cellular function by affecting ion channels. ROS were shown to regulate various types of ion channels, such as Ca2+-dependent K+ channels [8,9], ATP-sensitive K+ channel [9,10], HERG channels [11], and Ca2+ channels [12,13], and also affected Ca2+ release-activated Ca2+ current [14]. In addition, ROS exert complex effects on voltage-dependent K+ channels (Kvs). Hydrogen peroxide significantly accelerated the activation kinetics of Kv1.4 and Kv3.4, whereas did not alter Kv1.3, Kv2.1 and Kv2.2 [15]. In addition, hydrogen peroxide negatively shifted the activation curve of Kv1.5 [16]. Conversely, superoxide decreased the strength of the current through this channel [17]. Ca2+-dependent K+ channels were activated by hydrogen peroxide [8] or superoxide [9] radicals, resulting in the hyperpolarization of the membrane, or dilation of cerebral arterioles. Both superoxide and hydrogen peroxide radicals enhanced the activity of large-conductance Ca2+-dependent K+ channel in rat and cat cerebral arterioles; in contrast, the peroxynitrite radical decreased this activity in rat cerebral arteries [18].
Evidence indicates that KCa3.1 profoundly regulates endothelial function. It mediates a part of the endothelium-derived hyperpolarization response, and contributes to endothelium-dependent relaxation of blood vessels. In addition, KCa3.1 modulates Ca2+ influx in endothelial cells. On inhibition of KCa3.1, Ca2+ influx and endothelium-dependent relaxation effects were abrogated [19]. Thus, KCa3.1 dysregulation causes endothelial dysfunction, and thereby may contribute to vascular diseases such as preeclampsia [20] and Fabry disease [21]. In KCa3.1-deficient mice, the endothelial KCa3.1 current was abolished, leading to a considerable increase in arterial blood pressure and to a mild left ventricular hypertrophy [22]. We previously reported that in α-galactosidase A-knockout mice (an animal model of Fabry disease), endothelial dysfunction is caused by KCa3.1 downregulation and dysfunction [21]. Furthermore, we suggested that KCa3.1 downregulation contributes to the endothelial dysfunction seen in preeclampsia [20]. However, the role of ROS in the regulation of endothelial KCa3.1 has received little attention.
In the present study, we compared the effects of hydrogen peroxide and superoxide radicals on KCa3.1 expression in human umbilical vein endothelial cells (HUVECs). We found that the hydrogen peroxide radical donor TBHP increased KCa3.1 expression by upregulating pERK expression and downregulating repressor element-1 silencing transcription factor (REST) expression, and augmented KCa3.1 current. On the other hand, the superoxide donor, xanthine/xanthine oxidase mixture (X/XO), or L-α-lysophosphatidylcholine (LPC) decreased KCa3.1 expression by downregulating pERK expression and upregulating REST expression, and inhibited KCa3.1 current.
Go to : 
METHODS
Cell culture
Endothelial cells were isolated from human umbilical veins by collagenase treatment, as previously described [23]. HUVECs in suspension were plated into 6 well cell culture plates and grown in complete M199 (Hyclone, Logan, UT) supplemented with 10% fetal bovine serum (FBS; Life Technologies Corp., Carlsbad, CA), 100 units/ml penicillin, 100 µg/ml streptomycin, 15 µg/ml endothelial cell growth supplement (BD Biosciences, Rockville, MD), 0.1 mM MEM non-essential amino acids (Life Technologies Corp., Carlsbad, CA) and 10 unit/ml heparin. Cultured cells were identified as endothelial cells in origin by their cobble stone appearance at confluence and positive staining with 1,1'-dioctadecyl-3,3,3',3'-tetramethyl-indocarbocyanine perchlorate-labelled acetylated low density lipoprotein (Biomedical Technologies Inc., Stoughton, MA). HUVECs were used up to the second or third passage.
The investigation was approved by local ethics committee, the Institutional Review Board of the Ewha Womans University, and was in accordance with the Declaration of Helsinki.
Real-time PCR and immunoblot analysis
RNA isolation was performed using the RNeasy Mini Kit (Qiagen, Valencia, CA), and RNA was then reverse transcribed using a High Capacity cDNA Archive Kit (Applied Biosystems, Foster City, CA). PCRs were performed on an ABI 7000 sequence detection system (Applied Biosystems) using a SYBR Green PCR Master Mix (Applied Biosystems). Primers for REST were 5'-GTGCGAACTCACACAGGAGA-3' (sense) and 5'-AAGAGGTTTAGGCCCGTTGT-3' (antisense). mRNA expression was normalized to the house-keeping gene, human Gapdh (5-GGCCTCCAAGGAGTAAGACC-3' (sense) and 5'-AGGGGTCTACATGGCAACTG-3' (antisense)).
For immunoblot analysis, 30 µg of protein from cell homogenates was subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (7.5~12% gels), and proteins were then transferred to a nitrocellulose membrane. Membranes were blocked for 1 hour with TBST (10 mM Tris-HCl, 150 mM NaCl, and 1% Tween 20, pH 7.6) containing 5% bovine serum albumin at room temperature. The blots were incubated for 3 hours with primary antibody against primary KCa3.1 antibody (IK1; Santa Cruz Biotechnology, Santa Cruz, CA), or p-ERK (Cell Signaling Technology, Beverly, MA), followed by incubation with horseradish peroxidase-conjugated secondary antibodies for 1 hour. Bands were visualized by chemiluminescence. Data collection and processing were performed using a luminescent image analyzer LAS-3000 and Image Gauge software (FujiFilm, Tokyo, Japan).
Immunocytochemistry
HUVECs were grown on glass coverslips precoated with 1% gelatin. Cells were incubated overnight at 4℃ with a diluted (1:50) primary KCa3.1 antibody, washed and incubated for 1 hour at room temperature with a secondary antibody, Alexa Flour 488 donkey anti-goat IgG (1:1,500; Molecular probes, Eugene, OR). After that, the cells were counterstained with 4',6-diamidino-2-phenylindole dihydrochloride (DAPI). The mounted coverslips were viewed under a confocal microscope (Carl Zeiss, Gottingen, Germany) and photographed as described previously [24].
Electrophysiology
The patch-clamp technique was used in whole-cell configurations at room temperature. Whole-cell currents were measured using ruptured patches and monitored in voltage-clamp modes with an EPC-9 (HEKA Elektronik, Lambrecht, Germany). The holding potential was 0 mV and currents were monitored by the repetitive application of voltage ramps from -100 to +100 mV with a 10-second interval (sampling interval 0.5 milliseconds, 650 millisecond duration). The standard external solution contained (in mM): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 HEPES, 10 glucose, pH adjusted to 7.4 with NaOH. The pipette solution for whole-cell recording contained (in mM): 40 KCl, 100 K-aspartate, 2 MgCl2, 0.1 EGTA, 4 Na2ATP, 10 HEPES, pH adjusted to 7.2 with KOH. For buffering free Ca2+, the appropriate amount of Ca2+ (calculated using CaBuf software; G. Droogmans, Leuven, Belgium; ftp://ftp.cc.kuleuven.ac.be/pub/droogmans/cabuf.zip) was added in the presence of 5 mM EGTA.
KCa3.1 current was activated by loading 1 µM Ca2+ via a patch pipette in whole-cell clamped HUVECs and adding the KCa3.1 activator 1-ethyl-2-benzimidazolinone (1-EBIO, 100 µM) to the external solution. KCa3.1 current was normalized to cell capacitance and the selective KCa3.1 blocker TRAM-34-sensitive current was measured as KCa3.1 current.
Chemicals
LPC from egg yolk, N-acetyl-L-cysteine (NAC), superoxide dismutase (SOD), TBHP, tempol, tiron and TRAM-34 were purchased from Sigma-Aldrich (St. Louis, MO); DAPI from Molecular Probes (Eugene, OR), X/XO from Calbiochem (Gibbstown, NJ), 1-EBIO from Tocris Bioscience (Ellisville, MO). TRAM-34 and 1-EBIO were applied to the bath solution at 10 µM and 100 µM, respectively. LPC, NAC, tempol, tiron, TRAM-34 and 1-EBIO were dissolved in DMSO. The final concentration of DMSO was less than 0.05%.
Statistical analysis
Pooled data are given as mean±SEM. Statistical evaluation of data was performed by Student t test. Values of p <0.05 were considered significant.
Go to :
RESULTS
Effect of ROS donors on endothelial KCa3.1 expression
We examined whether the ROS donors, TBHP or X/XO, can modulate KCa3.1 expression in endothelial cells. Primary HUVECs were incubated with ROS donors or vehicle for 24 h, followed by western blot analysis of protein expression (Fig. 1). Treatment with the hydrogen peroxide donor TBHP, but not the vehicle, unregulated KCa3.1 expression in a concentration-dependent manner (Fig. 1A). In contrast, treatment with the superoxide donor X/XO, but not the vehicle, downregulated KCa3.1 expression in a concentration-dependent manner (Fig. 1B). In addition, we examined the effects of LPC, a molecule involved in superoxide generation in endothelial cells [3], on KCa3.1 expression (Fig. 1C). Similar to the X/XO effect, treatment with LPC decreased KCa3.1 expression in a concentration-dependent manner. To corroborate these findings we next examined whether ROS donors affected KCa3.1 expression in HUVECs by using immunocytochemical staining to detect expression of KCa3.1 protein (Fig. 2). KCa3.1 expression (indicated by green florescence) was markedly increased in HUVECs treated with TBHP compared to those treated with vehicle (Fig. 2A). In contrast, KCa3.1 expression was markedly lower in HUVECs treated with X/XO than in those treated with vehicle (Fig. 2B). These results, which indicate that KCa3.1 expression is upregulated or downregulated following treatment with TBHP and X/XO, respectively, suggest that the hydrogen peroxide and superoxide radicals exert converse effects on the expression of this ion channel.
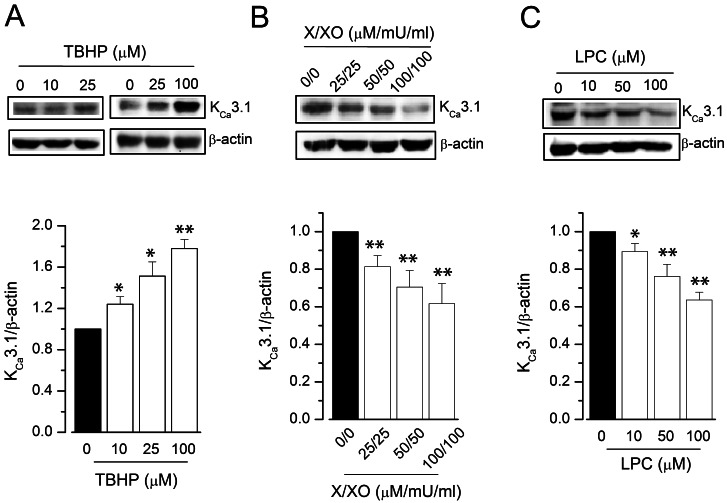 | Fig. 1KCa3.1 expression in ROS donor-treated HUVECs. KCa3.1 expression was measured using immunoblot, and relative protein expression was represented as a ratio of the levels in the vehicle-treated group to that in the test group. HUVECs were treated with TBHP (A), X/XO (B), or LPC (C), for 24 h; n=4~7. *p<0.05, **p<0.01 versus vehicle-treated control.
|
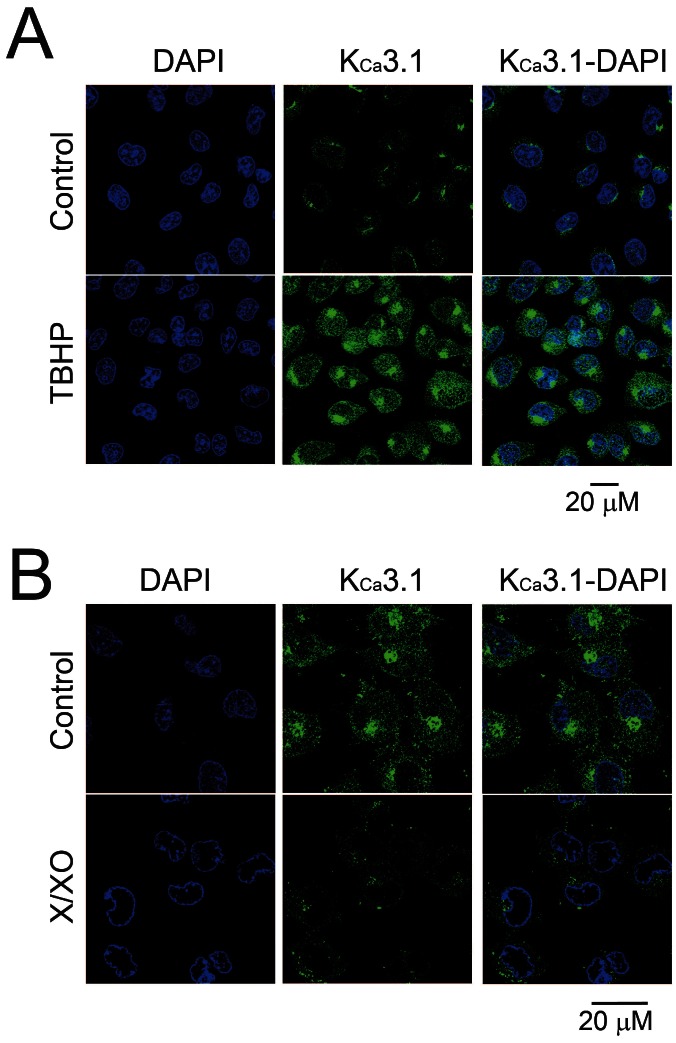 | Fig. 2KCa3.1 expression in ROS donor-treated HUVECs. KCa3.1 expressions were measured using immunocytochemistry. HUVECs were treated with 50 µM TBHP (A), or 50 µM/50 mU/ml X/XO (B), for 24 h. Immunocytochemistry images show KCa3.1 staining in green, and nuclear staining with DAPI in blue; combined images are also presented.
|
Antioxidants prevent ROS donor-induced modulation of KCa3.1 expression
We then examined whether treatment with antioxidants or SOD could prevent the ROS donor- or LPC-induced modulation of KCa3.1 expression. Pre-treatment of HUVECs with the antioxidants tempol, tiron, or NAC prevented TBHP-induced upregulation (Fig. 3A), as well as X/XO-induced downregulation (Fig. 3B) of KCa3.1 expression. Furthermore, LPC-induced KCa3.1 downregulation was abrogated by pre-treatment with SOD (Fig. 3B). In combination, these findings reinforce that KCa3.1 is upregulated by the hydrogen peroxide radical, and downregulated by the superoxide radical.
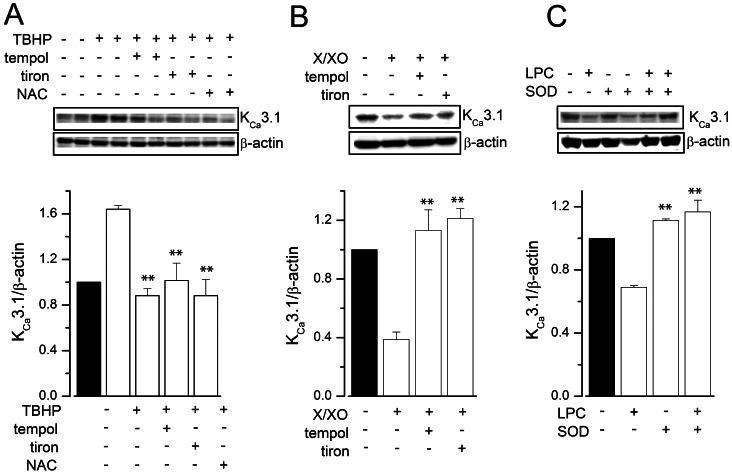 | Fig. 3Effects of antioxidants on ROS donor-induced KCa3.1 regulation. HUVECs were treated with vehicle or 100 µM TBHP (A), 100 µM/100 mU/ml X/XO (B), or 100 µM LPC (C), for 24 h, with or without pre-treatment with antioxidants (5 µM tempol, 50 µM tiron, or 10 mM NAC), or SOD (1,000 U/ml). The results are mean±SEM of three independent experiments. **p<0.01 versus TBHP, X/XO or LPC alone.
|
Effect of ROS donors on pERK and REST
To further evaluate the molecular mechanisms of ROS donor-induced modulation of KCa3.1 expression, we examined whether ROS donors affected the signal transduction pathways involved in KCa3.1 expression, the ERK pathway [25] and REST [26]. We thus assessed the phosphorylation of ERK (pERK) by immunoblot analysis (Fig. 4). TBHP treatment for 24 h significantly increased the levels of pERK in HUVECs (34±4% and 145±30% increase by 25 and 100 µM TBHP, respectively; Fig. 4A). In contrast, pERK levels were significantly reduced in HUVECs treated with X/XO (25 µM/25 mU/ml) for 24 h (57±4% inhibition; Fig. 4B). In addition, LPC reduced the level of pERK in a concentration-dependent manner in HUVECs (Fig. 4C). We then examined whether ROS donors modulate KCa3.1 expression by regulating the REST pathway. The expression of this transcription factor in HUVECs was significantly downregulated after incubation with TBHP in a concentration-dependent manner (29±3% decrease by 25 µM TBHP; Fig. 5A). In contrast, X/XO significantly upregulated REST expression in HUVECs (83±16% increase by 100 µM/100 mU/ml X/XO; Fig. 5B). These data suggest that TBHP upregulates KCa3.1 by activating the ERK pathway and downregulating REST. Conversely, X/XO downregulates KCa3.1 by inhibiting the ERK pathway and increasing REST expression.
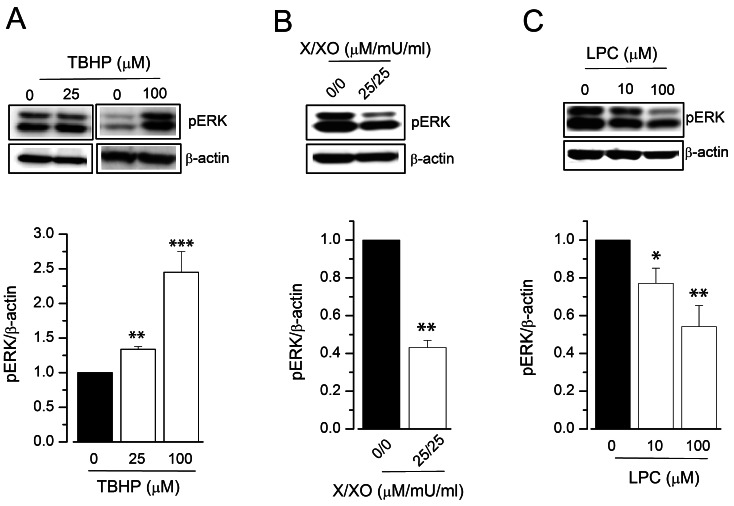 | Fig. 4Effect of ROS donors on pERK expression in HUVECs. pERK expression was measured using immunoblot, and relative protein expression was expressed as a ratio of the levels in the vehicle-treated group to that in the test group. HUVECs were treated with TBHP (A), X/XO (B), or LPC (C), for 24 h; n=4~7. *p<0.05, **p<0.01, ***p<0.005 versus vehicle-treated control.
|
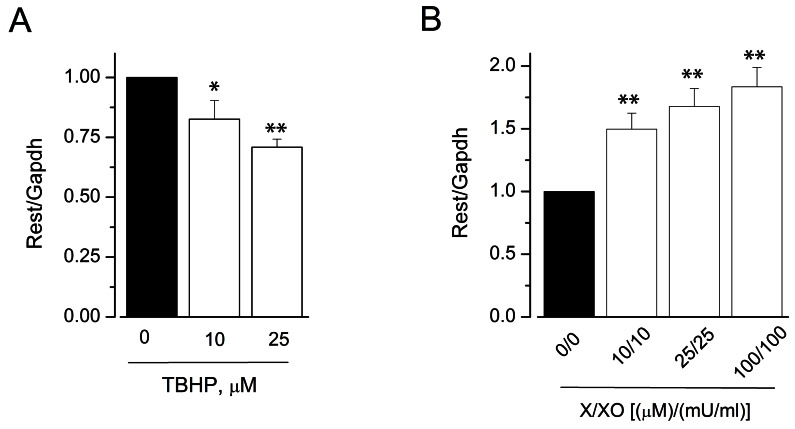 | Fig. 5Effect of ROS donors on REST expression in HUVECs. REST expression was measured using real-time PCR and the relative expression of mRNA was expressed as a ratio of the levels in the vehicle-treated group to that in the test group. HUVECs were treated with TBHP (A) or X/XO (B) for 24 h; n=4~7. *p<0.05, **p<0.01 versus control.
|
ROS donors modulate KCa3.1 currents in endothelial cells
We next examined whether the ROS donors could modulate KCa3.1 current, which was activated by loading whole cell-clamped HUVECs with 1 µM Ca2+, via a patch pipette, and supplementing the medium with the KCa3.1 activator 1-EBIO (100 µM). When the KCa3.1 current reached a steady state, TBHP or X/XO was applied in the external solution. The activated KCa3.1 current was further enhanced by TBHP (Fig. 6A, C), but it was inhibited by X/XO (Fig. 6D, F). The effect of TBHP (Fig. 6B, C), or X/XO (Fig. 6E, F) was inhibited by tempol. These data suggest that KCa3.1 current is augmented by the hydrogen peroxide radical, and inhibited by the superoxide radical.
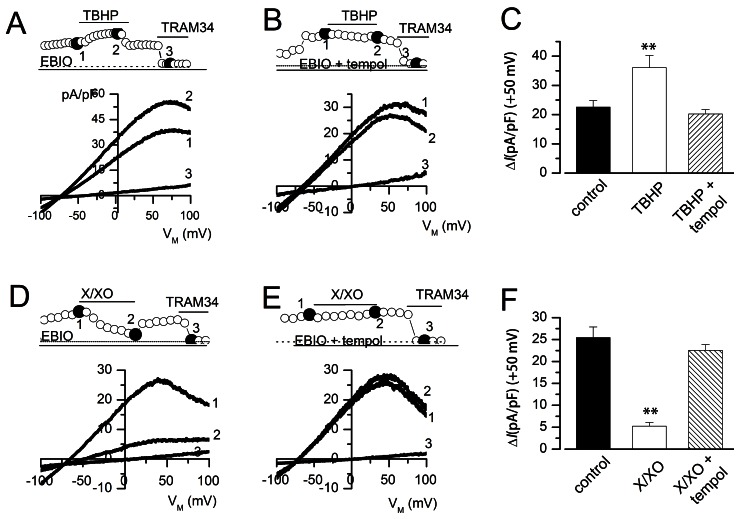 | Fig. 6Effect of ROS donors on KCa3.1 current in HUVECs. KCa3.1 current was activated by loading cells with 1 µM Ca2+ by using a patch pipette, and treating the cells with 1-EBIO (100 µM). The KCa3.1 currents were normalized to cell capacitance, and the TRAM-34-sensitive current was measured as the KCa3.1 current. (A, B, D, F) Current densities are shown at a membrane potential of +50 mV, and marked by open circles, while I/V relationships were obtained at the points marked by closed circles. (C, F) KCa3.1 current densities at +50 mV; n=6~8 (right panel). *p<0.05 versus control.
|
Go to :
DISCUSSION
In this study, we show that ROS regulate KCa3.1 expression through the modulation of the ERK and REST pathways, and influence KCa3.1 current production in human endothelial cells. The hydrogen peroxide donor TBHP increases KCa3.1 expression through pERK upregulation and REST downregulation. In contrast, the superoxide donors X/XO and LPC decrease KCa3.1 expression through pERK downregulation and REST upregulation. In addition, KCa3.1 current is augmented by TBHP, and inhibited by X/XO. These findings shed light on the mechanisms under-lying ROS-mediated regulation of this ion channel, a process that may be implicated in ROS-induced modulation of endothelial function.
Furthermore, the effects of the ROS donors, TBHP, XXO, or LPC were nullified by pre-treatment with antioxidants. The upregulation in KCa3.1 expression by the hydrogen peroxide donor TBHP was abrogated by antioxidants, suggesting that this effect is mediated by the hydrogen peroxide radical. In parallel, antioxidant treatment also nullified the ion-channel downregulating effects of the superoxide radical donors X/XO or LPC. LPC suppressed SOD1 and increased catalase expression [3], indicating that LPC increases the production of the superoxide, but not the hydrogen peroxide radical. These results suggest that X/XO- or LPC-induced downregulation of KCa3.1 is mediated by the superoxide radical. Since the ion channel KCa3.1 is an important physiological modulator in the endothelium, these findings suggest that ROS may affect endothelial function by regulating KCa3.1 expression and function.
Superoxide and hydrogen peroxide radicals are generated in vascular endothelial cells by several cellular enzymes, including eNOS, cytochrome P-450, and NADPH oxidases [27]. eNOS was suggested to contribute to ROS production in the endothelium, since acetylcholine-induced hydrogen peroxide radical production was markedly reduced in the blood vessels of eNOS-knockout mice [5], and eNOS activation generates superoxide radicals under the depletion of tetrahydrobiopterin [28,29]. In addition, cytochrome P-450 may be a source of ROS, since the hydrogen peroxide radical was produced in a cytochrome P-450-dependent manner in the coronary arteries of rats [30] and pigs [31]. NADPH oxidases play an important role in the generation of endothelial superoxide radicals. The enzymes are regulated by various stimuli such as shear stress, hypoxia, and hyper-lipidemia [2]. Since the production of superoxide radicals promotes generation of hydrogen peroxide radicals through SOD, both free radicals may be produced in endothelial cells in response to various stimuli, and may thereby regulate KCa3.1 expression and its function.
KCa3.1 expression is upregulated by the ERK pathway [25] and downregulated by REST [26]. External stress and physiological stimuli can affect the MAPK pathway by generating ROS [32]. Blood flow regulates KCa3.1 expression; a laminar flow upregulates KCa3.1 by increasing hydrogen peroxide generation, whereas turbulent flow downregulates KCa3.1 by increasing superoxide generation [33,34]. ERK phosphorylation and eNOS expression were inhibited by superoxide donors, but stimulated by the hydrogen peroxide radical donor THBP [3]. Thus, an inhibition of ERK phosphorylation may suppress KCa3.1 expression, as suggested previously [25]. In addition, ROS can activate as well as inactivate transcription factors [35]. The apparent regulation of REST levels and activity by transcriptional and post-transcriptional mechanisms [36] suggest that ROS are likely to regulate REST by affecting transcription factors. In addition, REST can modulate the phosphoinositide-3 kinase-Akt/ERK pathway, since cells lacking REST exhibit increased phosphoinositide-3 kinase signalling [37].
There are many reports on the superoxide or hydrogen peroxide radical-induced modulation of K+ currents, such as KCa1.1 current [38-40], the voltage-dependent K+ current [16,17], and the ATP-sensitive K+ current [10,38]. However, this is the first report of the superoxide or hydrogen peroxide radical-induced modulation of the current generated through the ion channel KCa3.1. We observed that the hydrogen peroxide radical stimulates KCa3.1 current in cells dialyzed with 5 mM EGTA. It is possible that a hydrogen peroxide radical-induced increase in cytosolic free Ca2+ [41,42] could lead to activation of KCa current. However, a high intracellular EGTA concentration may buffer the increase in Ca2+ induced by ROS. Since KCa3.1 current is activated in a voltage- and cytosolic free Ca2+-dependent manner, the augmentation of KCa3.1 current by the hydrogen peroxide radical suggests that this radical increases the sensitivity of KCa3.1 channel to voltage, or cytosolic free Ca2+.
In conclusion, the ROS donors TBHP, X/XO, and LPC regulate KCa3.1 expression in HUVECs. In addition, KCa3.1 current is modulated by the ROS donors TBHP, and X/XO. These ROS donor effects on KCa3.1 expression and the K+ current are prevented by pre-treatment with antioxidants. KCa3.1 plays an important role in vasomotor regulation; therefore, modulation of this ion channel in the vascular endothelium may be useful in the treatment of atherosclerosis and endothelial damage.
Go to :
 XML Download
XML Download