

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
CaSR
calcium sensing receptor
CCE
capacitive Ca2+ entry
CH
chronic hypoxia
CO
cardiac output
ET-1
endothelin-1
HIF-1
hypoxic inducible factor-1
HPV
hypoxic pulmonary vasoconstriction
5-HT
5-hydroxytryptamine
HVA
high voltagegated dihydropyridine-sensitive L-type channels
IP3
inositol triphosphate
IPAH
idiopathic pulmonary arterial hypertension
LVA
low voltage-gated T-type channels
miR
micro RNAs
mPAP
mean pulmonary artery pressure
NFAT
nuclear factor of activated T cells
NOS
nitric oxide synthase
PAEC
pulmonary artery endothelial cells
PASMC
pulmonary arterial smooth muscle cell
PGI2
prostaglandin
PH
pulmonary hypertension
PLC
phospholipase C
PPAR
peroxisome proliferator activated receptor
PVR
pulmonary vascular resistance
SOCE
store operated calcium entry
STIM1
stromal interaction molecule 1
TRP
transient receptor potential
VDCC
voltage dependent Ca2+ channels
INTRODUCTION
Calcium signaling impacts almost every aspect of cellular existence. It is the most common second messenger and, as such, regulation of calcium homeostasis within cells can have pertinent effects on cellular function. Pulmonary Hypertension (PH) is just one disease where changes in intracellular free calcium concentration ([Ca2+]cyt) can have significant impact in defining the pathogenic mechanisms leading to its development and persistence.
PH is a rare but severe and fatal lung disease, affecting predominantly women, which is caused by a plethora of mechanisms. The latest WHO clinical classification of PH groups manifestations of disease with similarities in the pathophysiologic mechanisms, clinical presentation, and therapeutic approaches together [1]. The pulmonary artery under normal conditions is maintained as a low resistance low pressure system which enables it to receive the entire cardiac output (CO) at one time. Mean pulmonary artery pressure (mPAP) is normally maintained at 16 mmHg. PH is characterized by an increase in mPAP to ≥25 mmHg at rest. Such an elevation of mPAP is typically due to increased pulmonary vascular resistance (PVR) as determined by the equation PVR×CO=mPAP. When blood flow through the pulmonary artery becomes restricted the right side of the heart compensates by pumping more forcefully, when this is sustained the right hand side of the heart becomes increasingly muscular and right ventricular hypertrophy ensues, the prognosis is poor. One of the hallmarks of PH contributing to the increase in PVR is a severe obstructive vasculopathy where the intima, media and adventitia are significantly thickened and more distal vessels become increasingly muscular. Characteristic vascular lesions, such as plexiform lesions and neointimal proliferation, also lead to obstruction of the pulmonary arteries [2]. Vasoconstriction of the arteries or occlusion due to in situ thrombosis is other examples contributing to the increase PVR in PH.
There has been a whole plethora of mechanisms associated with the development and progression of PH. Such complexity makes it difficult to isolate one particular pathway to target clinically. One commonality amongst these deregulated signaling pathways is the elevation of [Ca2+]cyt contributing to pulmonary vasoconstriction and excessive proliferation of smooth muscle cells and ultimately pulmonary vascular remodeling. The topic of calcium regulation in PH has been widely studied and there are a number of comprehensive reviews which I direct readers to [3-5]. Current therapeutic approaches, for example prostacyclin derivatives, endothelin-receptor antagonists, and phosphodiesterase type 5 inhibitors, have been unable to substantially decrease the morbidity and mortality due to PH. New mechanisms and novel therapeutic targets in PH are still at the forefront of research into PH and the current review serves to summarize the some of the most recent advances in the regulation of calcium during pulmonary hypertension.
Go to : 
VOLTAGE-DEPENDENT CALCIUM CHANNELS
Spanning the cell membrane are assortments of channels each allowing the specific transport of ions in or out of the cells. Voltage dependent Ca2+ channels (VDCC) are four domains, 6 transmembrane spanning proteins which have been functionally classified by their activation voltages. Low voltage-gated T-type channels (LVA) and high voltage-gated dihydropyridine-sensitive L-type channels (HVA) have both been identified with electrophysiological data supporting a functional role in the pulmonary artery, reviewed in Firth et al. [6]. The channels comprise of pore forming α subunits and additional regulatory subunits (β, α2δ and γ). Despite the detection of six α1 subunits at the transcriptional level functional evidence suggests that channels are either encoded by the α1c-subunit (L-type VDCC) or the α1G-subunit (T-type VDCC). L-type calcium channels are widely accepted as the source for depolarization dependent Ca2+ influx in pulmonary arterial smooth muscle cell (PASMC). The activity of these channels is largely controlled by membrane potential and voltage-gated potassium channels (Kv channels) are proposed to be the major regulators of resting membrane potential in PASMC. Inhibition of Kv channel expression and function is described in PASMC exposed to chronic hypoxia (CH) and those isolated from patients with idiopathic pulmonary arterial hypertension (IPAH); this change in Kv current is sufficient to depolarize the membrane and activate L-type VDCC Ca2+ influx [7-11].
T-type calcium channels have recently emerged as potential targets in PH. They are low voltage activated channels encoded by the Ca(v)3 family of genes which have been shown to be key source for Ca2+ influx to regulate cell cycle progression and, therefore, in the regulation of PASMC proliferation [12,13]. In normal PASMC, the Ca(v)3.1 isoform has been identified and its inhibition prevented entry into the cell cycle preventing a proliferative response [13]. Ca(v)3.1 has been specifically linked to the expression and activation of cyclin D further supporting its importance in regulating cell cycle suppressing [14]. In pulmonary artery endothelial cells (PAEC), isolated from the CH induced experimental model of PH, a decreased ATP-dependent and depolarization induced Ca2+ entry via mibefradil-sensitive T-type channels has been observed [15]. Such function regulation would imply a potential role in PH and in particular in pulmonary vascular remodeling. It will be important to fully explore the regulation of T-type channels in experimental models of PH and in human disease cells.
Go to :
STORE OPERATED CALCIUM ENTRY (SOCE): TRP, STIM AND ORAI
Hypoxic pulmonary vasoconstriction (HPV) is one of the first responses adopted by the pulmonary vasculature in response to decreased partial pressure of oxygen. After sensing a decreased oxygen tension the pulmonary arteries constrict to divert the blood flow to match oxygen tension to perfusion. When HPV is sustained it can lead to more permanent changes in the pulmonary vasculature such as pulmonary vascular remodeling and the development of PH. While it is known that HPV is mediated by decreased K+ channel currents causing depolarization activated Ca2+ influx through voltage-gated channels there was an uncertainty as to why HPV was reliant upon a high degree of pre-constriction in isolated rat pulmonary artery. Back in 2000, Robertson et al. investigated the involvement of intracellular stores [16]. These studies were amongst the first to show voltage dependent and voltage independent phases contributing to HPV. The voltage independent phase was contingent upon a store depletion mediated capacitive Ca2+ entry (CCE) [16]. Since these observations there has been a distinct focus on identifying the molecular correlates for this store operated calcium entry (SOCE) pathway.
The precise identity of the channels constituting SOCE in PASMC has been debated over recent years, however evidence suggests that transient receptor potential channels (TRP), stromal interaction molecule 1 (STIM1) and Orai (a fundamental Ca2+ release activated Ca2+ channel pore-forming subunit in the plasma membrane) may act in concert or independently to drive SOCE. Responses to hypoxia are greater in distal PASMC over more proximally isolated cells. Using a combination of Ca2+-free extracellular solutions and cyclopiazonic acid to deplete the endoplasmic/sarcoplasmic reticulum (ER/SR) stores of Ca2+ Lu et al. were able to show that SOCE is important in HPV and that the response of the more distal pulmonary artery is greater most likely due to the increased SOCE [17]. Although the identity of the SOCE channel was not determined, expression of STIM1 and TRPC1, 3, 4, and 6 isoforms were detected at higher protein and mRNA levels in the distal pulmonary artery. TRP are a family of non-selective ion channels that are known to encode the store-operated Ca2+ channels (SOC) activated by Ca2+ store depletion (reviewed by [18]). CCE via TRP channels is thought to be important in human PASMC proliferation [19,20] and in enhanced [Ca2+]cyt during exposure to chronic hypoxia [21] and in a hypoxia inducible factor-1 (HIF-1) dependent manner [22,23]. TRPC6 is also known to be upregulated in PASMC from IPAH patients [24]. STIM1 acts as a sensor of ER/SR Ca2+ concentration. At rest it is diffusely present in the ER/SR membrane, upon depletion of Ca2+ from the ER/SR, it oligomerizes and translocates to discrete punctae in the ER/SR membrane that are in close proximity to the plasma membrane [25]. Using a fluorescence resonance energy transfer (FRET) technique, Navarro-Borelly and colleagues demonstrated a direct redistribution and interaction of both STIM-1 and Orai-1 in response to store depletion of Ca2+ [26]. Around the same time Liao et al. used a combination of electrophysiological techniques and intracellular Ca2+ imaging to demonstrate a similar STIM-1 interaction between both Orai-1 and TRPC subunits [27]. These observations have also been demonstrated in mouse pulmonary artery smooth muscle cells where Ng and colleagues showed co-immunoprecipitation of TRPC1 with STIM1 and of Orai with STIM-1 [28,29]. Post store depletion the precipitation level and co-localization of STIM-1 and Orai increased [28,29].
SOCE is shown to be a feature of several other known proponents of pulmonary hypertension, for example endothelin-1 (ET-1) and platelet derived growth factor (PDGF) mediated signaling. ET-1 production is enhanced and expression of its receptors upregulated in PH [30-32]. ET-1 induced pulmonary vasoconstriction of monocrotaline treated rats is partially inhibited by SOCE blockers including gadolinium (Gd3+), lanthanum (La3+), SKF-96365 and TRPC inhibitor BTP-2 [33]. PDGF is a potent mitogen which has been shown, along with its receptor, to be upregulated in models of PH and proposed to have a pertinent role in pulmonary vascular remodeling [34,35]. PDGF mediated PASMC proliferation is, in part, due to an upregulation of TRPC6 channels [36]. More recent data also linked PDGF with the activation of the Akt/mTOR pathway and, subsequently, to enhanced SOCE and cell proliferation in human PASMC. Inhibition of Akt attenuated the increase in [Ca2+]cyt and correlated with a significant downregulation of both STIM and Orai [37].
In addition to the TRPC channels, the TRPV channels have received some recent attention [38]. Stimulation of TRPV1 and V4 channels, identified in PASMC, leads to increased [Ca2+]cyt and PASMC migration with a correlating reorganization of the F-actin cytoskeleton and intermediate filament network [39]. Furthermore, TRPV4 appears to be important in the development of hypoxia-induced PH due to facilitated Ca2+ influx increasing pulmonary vasoconstriction and pulmonary vascular remodeling. This was supported by an enhanced myogenic tone and pulmonary vascular remodeling in hypoxic TRPV4 knockout mice [40]. The precise pathway linking TRP, STIM and Orai still remains to be fully elucidated; data does support an important role for all of the SOCE molecular correlates in the regulation of PASMC homeostasis and potentially implicated important roles in the development and pathogenesis of PH.
Go to :
CALCIUM-DEPENDENT REGULATION OF NFAT
As mentioned above a down regulation of Kv channels in PASMC and PA from patients with PH is now widely accepted. The associated membrane depolarization activate voltage dependent calcium channels leading to increased [Ca2+]cyt which has the knock on effects of contributing to increased contractility, enhanced cell proliferation and decreased cell apoptosis. It is, however still unclear what leads to the down regulation of these potassium channels. NFAT (nuclear factor of activated T cells) is a calcium/calcineurin-sensitive transcription factor which has recently been shown to be elevated in PASMC and circulating leukocytes in PH patients. NFAT isoform c2 inhibition did correlate to a restoration in Kv1.5 expression and function ultimately decreasing [Ca2+]cyt [41]. Another study using the chronic hypoxia mouse induced model of PH identified a requirement for NFAT isoform c3 [42]. CH induced endothelin-1 expression is a well-established phenomenon. In isolated mouse pulmonary resistance arteries NFATc3 was activated by endothelin-1, a response verified in human PASMC to involve Rho A kinase and actin polymerization [43]. A pathway whereby CH induced endothelin-1 expression enhances [Ca2+]cyt, Rho A kinase activity and actin polymerization (recently reviewed in [44,45]) leads to the activation of calcineurin, dephosphorylating NFATc3 and enhancing its translocation to the nucleus to become transcriptionally active can thus be implied from the current data (Fig. 1).
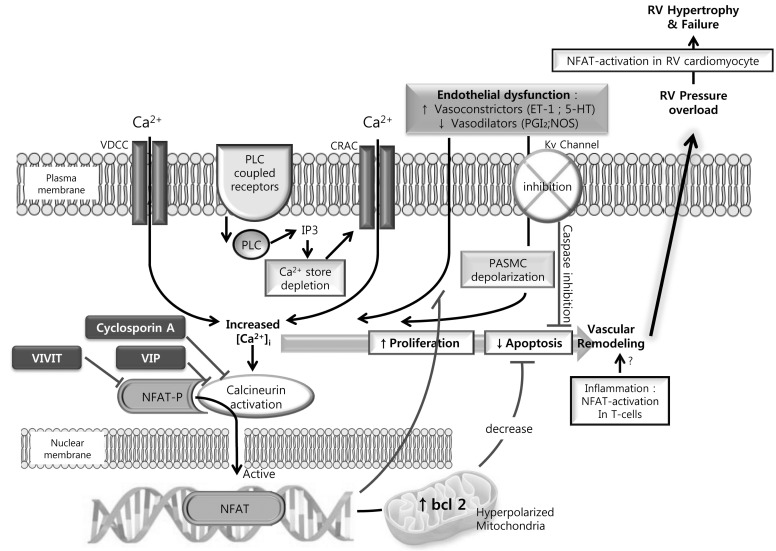 | Fig. 1The calcineurin-NFAT pathway as an integrator of multiple signaling pathways in the pathogenesis of pulmonary hypertension (PH). NFAT resides in the cytoplasm of resting cells in a phosphorylated and inactive state. Endothelial dysfunction occurs early in PH and results in an increased release of vasoconstrictors (Endothelin-1 [ET-1] and 5-Hydroxytryptamine [5-HT]) and decreased vasodilators (Prostaglandin [PGI2] and nitric oxide synthase [NOS]). These vasoconstrictors can stimulate phospholipase C (PLC) coupled cell surface receptors leading to mobilization of calcium ions (Ca2+) from intracellular stores via inositol trisphosphate (IP3). The elevated intracellular calcium ([Ca2+]i) can cause further Ca2+ influx via Ca2+ release-activated Ca2+ channels (CRAC). Addtionally, the down-regulation of Kv1.5 depolarized PASMC and will lead to the influx of via L-type voltage dependent Ca2+ channels (VDCC). The elevated [Ca2+]i activates phosphatase calcineurin which dephosphorylates NFAT allowing for its translocation to the nucleus. Here it is involved in the regulation of multiple genes. Multiple NFAT binding elements are present in the promoter regions of both the Kv1.5 and bcl-2 genes leading to a promotion of cell proliferation and suppressing mitochondrial-dependent apoptosis. Cyclosporin A inhibits calcineurin-ubstrate interactions and VIVITs electively inhibits NFAT activation.
|
It is interesting to again reflect on some of the similarities between mechanisms involved in the development of PH and of cancer. In mouse osteosarcoma FBJ-S1 and Lewis lung carcinoma cells an L-type VDCC/[Ca2+]cyt/calcineurin/NFAT signaling pathway has been shown to transcriptionally regulate the expression of caveolin-1 [46]. Caveolin-1 is already known to be an integral component in the pulmonary vascular remodeling in PH. Although data is somewhat conflicting between animal models and human cells it has been shown that its expression is increased in PASMC from patients with IPAH [47,48]. Caveolin-1 is an integral structural component of caveolae; a subset of membrane lipid rafts which serve as regions to coordinate cellular signaling. Thus targeting NFAT may be key to targeting several of the known pathways involved in the development and progression of PH.
Increased [Ca2+]cyt is a prerequisite for nuclear translocation of NFAT. As discussed above increased SOCE is now considered a key mechanism in the pathogenesis of PH. In addition to the entry of calcium via voltage dependent ion channels studies have shown that SOCE and CCE dependent increases in cytoplasmic calcium are directly linked to increased nuclear translocation of NFAT in the pulmonary vasculature [49,50]. Reports suggest that the anti-proliferative effects of sildenafil in PH are due to a mechanism in addition to the known NO/cGMP axis [51,52]. Wang and colleagues nicely demonstrated that the anti-proliferative effects of sildenafil in PASMC are due to the SOCE/[Ca2+]cyt/NFAT pathway [49]. Sildenafil successfully suppressed the hypoxia mediated increase in TRPC1 gene and protein levels and increased SOCE mediated nuclear translocation of NFAT in human PASMC increasing cell proliferation rates [49]. In calf PAEC NFATc1 translocates to the nucleus after elevation of [Ca2+]cyt by agonists like bradykinin or ATP. However, in the absence of extracellular calcium, CCE does not occur and translocation to the nucleus appears to be inhibited and therefore independent of Ca2+ release from the ER [50].
NFAT has been shown to crosstalk with both calcineurin and PPARγ; a role for PPAR in the pathogenesis of PH has become strikingly evident over the past decade. Peroxisome proliferator activated receptor (PPAR) is a member of the nuclear hormone receptor superfamily of ligand activated transcription factors [53]. Two isoforms exist differing in N terminal domains only but with distinct tissue distribution; isoform PPARγ2 is mostly associated with adipose tissue expression whereas the PPARγ1 is more widely expressed including brain, vascular tissues and lymphatic cells. In the lung expression has been shown in the pulmonary vasculature including both smooth muscle cells and endothelial cells, with decreased expression levels observed in PH patient derived cells and in in vivo animal models of PH [54]. In the case of cardiac hypertrophy, where this interaction has been more widely studied, elevation of PPARγ using ligands, such as rosiglitazone, has been shown to inhibit endothelin-1 mediated hypertrophy via NFAT/calcineurin signaling [55].
The PPAR family of transcription factors also includes two other isoforms: delta and beta. Like PPARγ they act by heterodimerizing with the RXR (retanoid X receptor) and then bind to peroxisome proliferator hormone response elements to regulate transcription of target genes. PPARβ/δ has been reported to modulate gene regulation in response to prostacyclin analogues like sildenafil. Like the sildenafil dependent regulation of SOCE/[Ca2+]cyt/NFAT pathway mentioned above, evidence for another NO/cGMP independent pathway of action exists: in this recent study acute prostacyclin-induced Ca2+-activated K+ channel activation in human PASMC was found to be reliant upon PPARβ/δ signaling [56].
Go to :
MICRORNA REGULATION
Micro RNAs (miR) were first identified in 1993 by Lee and colleagues [57]. It was not until the 2000's that they become more widely recognized as biological regulators with distinct and conserved functions. miR are short nucleotide sequences of ~22 nucleotide that act as post transcriptional regulators binding to the complementary sequence in the 3'UTR of target genes. Each miR is capable of targeting hundreds of genes with an estimated 60% of genes targeted. Over 700 miR are currently identified in humans with a predicted 800 to exist. Over the past 10 years miR has been shown to be both regulators of normal cell function and to be deregulated in disease. Their involvement in disease pathogenesis is seemingly endless. One of the most pertinent discoveries seems to be their ability to serve as biomarkers in, but not limited to; cancer [58-61], cardiovascular disease [62,63], multiple sclerosis [64], inflammatory bowel disease [65], schizophrenia [66] and rheumatoid arthritis [67].
It was only recently that a handful of miRs have been identified in the pulmonary vasculature with proposed pathophysiological roles in PH [68-70]. From these select few studies it is evident that the formation of plexiform lesions and pulmonary vascular remodeling involve regulation by miR activity [68-72]. Comparing plexiform lesions and concentric lesions dissected from the arteries of pulmonary arterial hypertension and control patients one study demonstrated that smooth muscle specific miRs 143 and 145 were significantly higher in concentric lesions; data supported by similar up-regulation of the miR target genes myocardin and smooth muscle heavy chain. miR-126, on the other hand, was augmented in plexiform lesions. VEGF-A is the major target of miR-126 and thus elevation of this miR enhances angiogenesis and thus helps to explain the pronounced angiogenic phenotype of these lesions [73,74]. In PASMC a correlating down-regulation of miR-204, acting by promotion of a STAT3 feedback loop leading to sustained activation of STAT3, was also observed supporting an enhanced cell proliferation [75]. Inhibition of the src/STAT3/Pim1 axis has been shown to improve monocrotaline-induced hypertension in rats by increasing apoptosis through depolarization of mitochondria and decreasing vessel contractility and proliferation due to decreased [Ca2+]cyt [76]. miR-328 is another miR identified in the pulmonary vasculature with a proposed role in chronic hypoxia mediated secondary pulmonary hypertension via regulation of L-Type calcium channels [70]. A miR-328 binding site in the 3'UTR of the L-Type Calcium channel isoform 1ac leads to an inhibition of its expression. In PH, miR-328 is significantly down-regulated leading to a concomitant up-regulation of L-Type calcium channels and thus an increased potential for elevation of [Ca2+]cyt [70]. In many essential ways the regulation of miR in pulmonary hypertension mirrors that in cancer with roles leading to increased cell proliferation and oncogenesis.
Go to :
CALCIUM SENSING RECEPTOR (CaSR)
A Calcium Sensing Receptor (CaSR) was first characterized and cloned from bovine parathyroid back in 1993 after a role in parathyroid hormone secretion was identified [77,78]. Soon after it was cloned and mapped to chromosome 3q13.3-21 in humans [79,80]. The CaSR is a 1078 amino acid encoded member of the G-protein-coupled receptor family. Its expression is most commonly associated with the parathyroid and kidney where it senses extracellular calcium levels to regulate parathyroid hormone (PTH) secretion and renal tubular calcium reabsorption in response to alterations in extracellular calcium. Since then its expression has been identified in a wide variety of tissues including, but not limited to; sensory nerves [81-83], pancreatic islet cells [84], osteoclasts [85,86], epithelial cells [87,88], hepatocytes [89,90], cardiomyocytes [91,92] and B cells [93]. Furthermore, mutations in the receptor have been identified and functionally linked to diseases like hyper- and hypo-calcemia, Bratter's Syndrome and hypoparathyroidism [94-98]. A variety of cancers have also been associated by an absence or loss of CaSR; pituitary adenomas and colorectal cancer [99-101]. In colorectal cancer a decreased CaSR expression in the colonic epithelium is evident; it is believed at CaSR acts to promote the down-regulation of beta-catenin-mediated transcriptional activation having subsequent effects on cell proliferation [87,102,103]. Functional expression is also identified in the cardiovascular system [91,104,105] and, most recently, in the pulmonary vasculature where it has been implicated in the pathogenesis of PH [106-109].
Given the importance of calcium in the development and pathogenesis of PH, it comes as no surprise that the CaSR is present in the pulmonary artery. Studies in isolated rat pulmonary arteries and PASMCs first identified functional CaSR [109]. This study demonstrated the presence of CaSR in PASMC and showed that it was involved in regulating pulmonary arterial tension by signaling through PLC and IP3 [109]. The same group followed this study by investigating how the CaSR was regulated in hypoxic condition; a pathway involving CaSR dependent MEK/ERK and PI3K activation contributed to the hypoxia induced increased proliferative rate of the PASMC [108]. While these studies implicated a role for CaSR in pulmonary vascular disease a more recent study by Yamamura and colleagues took IPAH patient cells and utilized the monocrotaline-induced animal model of PH to more specifically look at the role of CaSR in pulmonary disease [107]. In the IPAH cells, calcium influx was enhanced with calcimimetic R568 or decreased with calcylitic NPS2143 additionally, the expression of CaSR was significantly higher. More interestingly the development of PH and right ventricular hypertrophy in both monocrotaline-treated and hypoxia-exposed rats was prevented after injection of the calcilytic [107]. This study is really the first to show a pathogenic role for CaSR in PH and it will be interesting to see if CaSR promises to be a potent therapeutic target in patients with PH.
This review has highlighted some of the most recent advances in calcium signaling and regulation in PH. Hopefully, more studies will start to identify small molecules or other ways to manipulate the signaling pathways described and push more selective therapeutic approaches into the clinic.
Go to :
 XML Download
XML Download