

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The pancreatic β-cell is a specialized metabolic sensor of the body that releases insulin to maintain blood glucose levels in a narrow range. Glucose uptake elicits downstream signals accelerating the exocytosis of insulin granules in β-cell [1]. In this process, generation of ATP and other coupling factors from mitochondria play an important role [2]. Disturbing mitochondrial function in pancreatic β-cells impairs metabolism-secretion coupling and promotes the development of type 2 diabetes [2,3]. Mutations in mitochondrial DNA have been described that result in maternally inherited diabetes [4]. Furthermore, islet β-cells from diabetic patients display mitochondrial dysfunction [5].
Mitochondria are dynamic organelles, which continuously divide and fuse. These processes are mediated by fission and fusion proteins. The two main components of the fission machinery are Fis1 and Drp1 [6]. Drp1 translocates from the cytosol to predetermined fission sites on the mitochondria, and constricts the membrane by a GTPase-dependent mechanism [7]. Fis1 was suggested to recruit Drp1 to the mitochondrial outer membrane [8]. Mitochondrial fission factor has been reported as another Drp1 receptor in the fission process [9,10]. Inhibition of the fission proteins protects against apoptosis [11], but also impairs mitochondrial function by decreasing autophagocytosis [12].
Mitochondrial fusion is mediated by mitofusin 1 (Mfn1) and mitofusin 2 (Mfn2) in the outer membrane as well as Opa1 in the inner mitochondrial membrane [6]. Both Mfn1 and Mfn2 have a GTPase domain in the N-terminus and loss of function mutation in this domain disrupts fusion activity resulting in excessive fission when overexpressed [13]. Deletion of the Mfn1 or Mfn2 genes in the mouse results in mitochondrial dysfunction and embryonic lethality [14]. Mutations in Mfn2 cause a neurological disease affecting sensory and motor peripheral neurons [15].
Mitochondria continuously move in the cytosol, which is required for optimal cell function especially when the amount of the organelles is limiting [16]. This motility enables mitochondria to supply ATP and other metabolites even to distal parts of the cell or allows the organelle to buffer local Ca2+ increases efficiently [17]. Cytoskeletal tracks and several motor proteins responsible for mitochondrial movement have been identified [18]. Kinesin and dynein are involved in antegrade or retrograde movement of mitochondria along microtubules. Milton and Miro act as adaptors localized on mitochondria [17]. Miro is a Rho-GTPase with Ca2+ binding motifs [19]. This protein may mediate Ca2+-induced inhibition of mitochondrial motility in order to recruit active mitochondria to sites of local Ca2+ increase [20]. In pancreatic β-cells, mitochondrial localization and ATP supply to peripheral area might be important, because local rises of the ATP/ADP ratio are most likely required to induce the closure of plasmalemmal KATP channels leading to voltage-sensitive Ca2+ influx and insulin exocytosis [21].
We previously reported that mitochondrial fragmentation does not have a negative impact on mitochondrial function and glucose-stimulated insulin secretion in INS-1E cells [22]. On the other hand, overexpression of wild type mitofusin 1 (WT-Mfn1) evoked hyperfusion of the mitochondria with lowered cellular ATP levels, increased lactate production and impaired insulin release [22]. In this study, we report that WT-Mfn1 overexpression impairs mitochondrial motility and function in INS-1E cells as assessed by live cell confocal imaging and measurement of the mitochondrial membrane potential. These defects were not observed in cells overexpressing dominant negative mutant mitofusin 1 (DN-Mfn1).
METHODS
Cell culture
INS-1E rat clonal β-cells were cultured in a humidified atmosphere (37℃) containing 5% CO2 in complete medium composed of RPMI 1640 (Invitrogen, Carlsbad, CA, USA) supplemented with 10% FCS (Invitrogen), 1 mM sodium pyruvate, 50 µM 2-mercaptoethanol, 2 mM glutamine, 10 mM HEPES, 100 U/ml penicillin, and 100 µg/ml streptomycin.
Adenovirus infection
Doxycycline-inducible adenoviruses encoding WT-Mfn1 and DN-Mfn1 were constructed as described previously [22]. One day after plating INS-1E cells, adenoviruses encoding the transgene were applied together with the adenovirus expressing the reverse tetracycline-transactivator to INS-1E cells for 90~120 min 1 day after seeding [23]. Before the experiment, cells were cultured for 48 hours with or without doxycycline (500 ng/ml).
Mitochondrial morphology
Mitochondria of INS-1E cells were visualized by transfecting the mitochondrially-targeted red fluorescent protein (mitoRFP) plasmid using lipofectamine 2,000 (Invitrogen) followed by infection with adenoviruses. Mitochondrial morphology was observed 48 hrs after infection and analyzed using a confocal laser-scanning microscope (LSM510meta, Zeiss, Switzerland) after fixation with 4% paraformaldehyde.
Measurement of mitochondrial membrane potential
INS-1E cells were seeded (5×104 cells/well) on black-walled 96 wellplates and infected with adenoviruses. Cells were cultured in medium with or without doxycycline for 48 hours, and then loaded with 500 nM of JC-1 (Invitrogen) in KRBH buffer (2.8 mM glucose) for 30 minutes at 37℃. After washing, JC-1 fluorescence was measured from cells in KRBH buffer using a FlexStation (Molecular Devices Co., Sunnyvale, CA, USA) as described previously [22].
MTT assay
3-(4,5-dimethylhioazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) was purchased from Sigma (St. Louis, MO, USA). INS-1E cells seeded on 96-well microtiter plates were infected with different doses (30~120 ifu/cell) of adenovirus coding for WT-Mfn1 and incubated for 48 hours in medium containing 500 ng/ml doxycycline. Cells were further incubated with MTT (100 µg/well), and solubilized with DMSO. Absorbance of each well at 570 nm and 650 nm was measured using an ELISA reader (Molecular Devices Co.). The difference in absorption (A570-A650) was then calculated.
TUNEL assay
The fraction of apoptotic cells over-expressing WT-Mfn1 was determined using the TUNEL assay (In Situ Cell Death Detection Kit, Roche Diagnostics) according to the manufacturer's instructions. All nuclei were stained with DAPI (1 µg/ml for 5 minutes) and data were expressed as the percent of TUNEL positive cells per DAPI-stained cells.
Mitochondrial motility measurement
After transfecting mitoRFP, mitochondrial images were obtained using an inverted microscope (Zeiss, Switzerland) with confocal spinning disk (QLC100, VisiTech, UK). Image series were analyzed by Metamorph software (Universal Imaging, Molecular Devices Co.). The number of moved pixels per second were normalized to total pixels above the threshold (mitochondria) and used to calculate a 'motility index'.
RESULTS
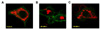
INS-1E cells were infected with adenoviruses encoding WT-Mfn1 or DN-Mfn1. Infection with these viruses in combination with reverse tetracycline-transactivator induced doxycycline-dependent expression in INS-1E cells as demonstrated previously [22]. To follow mitochondrial morphology and localization, we transiently transfected INS-1E cells with mitoRFP and plasmalemma-targeted GFP using lipofection. As shown in Fig. 1A, Mitochondria in control non-induced (without doxycycline incubation) cells showed a filamentous pattern of mitochondria distributed from the perinuclear area to the cell periphery. Overexpression of WT-Mfn1 caused aggregation of the mitochondria by promoting the fusion process. The mitochondria in WT-Mfn1-overexpressing cells did not reach the subplasmalemmal area (Fig. 1B). Conversely, DN-Mfn1 with a point mutation in the GTPase domain (T109A) caused fragmented mitochondria, which were distributed throughout the cytosol (Fig. 1C).
To assess mitochondrial function after manipulation of the fission/fusion machinery, we measured mitochondrial membrane potential (Δψm) changes using the fluorescent dye JC-1. This dye permits ratiometric recording of Δψm changes [22]. Glucose induces hyperpolarization of Δψm. Doxycycline (500 ng/ml) induction of WT-Mfn1 attenuated this hyperpolarization (Fig. 2A). Blockage of the F1F0-ATPase by oligomycin is known to elicit hyperpolarizing changes in well-functioning mitochondria [24]. Dysfunctional mitochondria maintain Δψm by reversal of the F1F0-ATPase, resulting in ATP consumption. Oligomycin in such dysfunctional mitochondria causes depolarization. In WT-Mfn1 overexpressing cells, oligomycin (2.5 µg/ml)-induced hyperpolarization of Δψm was strongly decreased when compared to control cells (Fig. 2A).
Mitochondrial aggregation and the depolarization of Δψm may cause cell death. To follow cell viability in cells overexpressing WT-Mfn1, we used the colorimetric MTT assay. The amount of formazan reaction product formed in the MTT assay reflects the mitochondrial enzyme activity, which allows assessing the cell viability. INS-1E cells were infected with increasing concentrations of WT-Mfn1. Even at the highest concentration of adenovirus particles (120 ifu/cell), WT-Mfn1 did not significantly altered cell viability (Fig. 2B). This result also implies that the mitochondrial enzyme activity at least involved in formazan reaction was not affected by WT-Mfn1 overexpression, although there was a functional deterioration of mitochondria. TUNEL assays further demonstrated that apoptosis was not induced by WT-Mfn1 overexpression (Fig. 2C). In contrast, Fis1 overexpression caused apoptotic cell death as reported previously [22].
We analyzed mitochondrial motility from series of confocal images (Fig. 3). Elongated mitochondria of control INS-1E cells displayed worm-like wriggling movements. Compared to control cells, WT-Mfn1 overexpressing cells had less motile, clumped mitochondria. Mitochondrial motion can be quantified by following changes of mitoRed positive areas. Fig. 3D~F are representative illustrations combining two consecutive images (red- and green-colored) after binarization. We measured the moved mitochondrial pixels and expressed as an arbitrary motility index as described in Materials and Methods. Consistent with our qualitative observations, mitochondrial aggregates after WT-Mfn1 overexpression had a much lower motility index than control cells (Fig. 4D). Conversely, fragmented mitochondria in DN-Mfn1 overexpressing cells showed fast random movement like Brownian motion. Microtubule-dependent movement was estimated by the application of nocodazole which interferes with polymerization of microtubules [20]. Nocodazole significantly attenuated the mitochondrial motility in control cells, but showed less inhibition in DN-Mfn1 overexpressing cells (Fig. 4E). FCCP, a mitochondrial uncoupler, elicited a further inhibition of motility maybe due to collapse of Δψm resulting in ATP depletion [25] or cytosolic Ca2+ increase [26]. Mitochondrial motility in WT-Mfn1 overexpressing cells was affected by neither nocodazole, nor FCCP.
DISCUSSION
Mitochondrial fusion is an essential process. Knockout mice deficient in either Mfn1 or Mfn2 display mid-gestational lethality [14]. In man, mutations in fusion genes can cause neurodegenerative diseases [15]. Fusion may be particularly important as it mediates the mixing of mitochondrial content where healthy mitochondria may be able to complement dysfunctional mitochondria [27]. Mitofusins also are known to have an impact on cell metabolism. Glucose oxidation, oxygen consumption, and mitochondrial membrane potential are lowered by loss of Mfn2 function [28]. Mfn2 tethers the ER to mitochondria, influences ER structure [29], regulates ER calcium homeostasis and store-operated calcium entry [30]. GTPase activity of Mfn1 and Mfn2 is an essential process for the fusion. Mfn1 GTPase activity is much higher than that of Mfn2 [31]. In this study, we overexpressed either wild type or GTPase-deficient Mfn1 and observed pronounced differences in mitochondrial morphology, dynamics, and function in INS-1E cells. Previously, we had demonstrated that WT-Mfn1 overexpression markedly reduced glucose-stimulated insulin secretion in this cell type [22]. The here presented data suggest that impaired metabolism-secretion coupling in WT-Mfn1 overexpressing cells is related to attenuated mitochondrial motility and function.
Hyperfusion of mitochondria due to WT-Mfn1 overexpression reduced glucose- and oligomycin-induced hyperpolarization. The extent to which mitochondria hyperpolarize in response to oligomycin reflects the quantity of functional mitochondria actively synthesizing ATP. Thus, overexpression of WT-Mfn1 increased the amount of dysfunctional mitochondria in the INS-1E cell population. These results explain our previous findings that INS-1E cells overexpressing WT-Mfn1 have reduced cellular ATP levels and enhanced lactate production [22]. One possible mechanism resulting in mitochondrial dysfunction is loss of mitochondrial autophagy (mitophagy) following Mfn1-induced mitochondrial aggregation. Mitophagy acts as a quality control mechanism by degrading dysfunctional mitochondrial fragments. Silencing of Fis1 or overexpression of dominant negative Drp1 deteriorates the ability of mitochondria to undergo mitophagy [12]. Compromised mitophagy causes the accumulation of oxidized mitochondrial proteins, reduces cellular respiration, and decreases glucose-stimulated insulin secretion [12]. Defects in mitophagy due to WT-Mfn1-induced hyperfusion may increase the proportion of dysfunctional mitochondria and therefore lower total mitochondrial activity.
For insulin secretion, mitochondrial movement to the cell periphery may be critical in order to supply ATP close to KATP channels to initiate β-cell electrical activity finally resulting in insulin granule exocytosis. To date, mitochondrial motility has been investigated mainly in neuronal cells which require mitochondria at sites distant from the cell body. We observed that INS-1E cells as well as pancreatic β-cells have tubular networks of filamentous mitochondria distributed throughout the cytosol [22]. In control INS-1E cells, the mitochondria were undergoing continuous wriggling movements and reached near plasma membrane areas of the cytosol. WT-Mfn1 overexpression caused aggregation and marked inhibition of the mitochondrial motility, preventing them from reaching the subplasma membrane areas. These findings are similar to those reported in neural cells with conditional knockout of Drp1 [32].
It has been reported that embryonic fibroblasts of mitofusin knockout mice have spherical highly fragmented mitochondria, which display disorganized random movement [14]. Similarly, mitochondria of INS-1E cells overexpressing DN-Mfn1 showed enhanced motility of fragmented mitochondria reaching even peripheral areas of the cell. In these cells, nocodazole inhibited mitochondrial movements less than in control cells. Overexpression of DN-Mfn1 may disrupt the association of the mitochondria to the microtubule system therefore allow random mitochondrial movement. Following DN-Mfn1 overexpression, mitochondria are more fragmented and possibly more likely to undergo mitophagy. In these cells, we reported that mitochondrial function was not reduced, but even enhanced [22]. Consistently, DN-Mfn1 neither increased apoptosis nor impaired metabolism-secretion coupling in these cells [22]. We suggest that fragmentation of mitochondria at least in some condition is beneficial in order for the cell to cope with various metabolic and/or oxidative stresses.
A large number of studies demonstrated that an imbalance of mitochondrial fission/fusion can secondarily also affect organelle function and motility. The exact roles of the mitochondrial-shaping proteins and their cell-type specific functions remain unclear. This study shows the negative impact of hyperfusion due to Mfn1 overexpression on mitochondrial activity. These defects likely explain impaired metabolism-secretion coupling in insulin-releasing cells overexpressing Mfn1. Further investigations to unravel the factors controlling mitochondrial morphology and motility are needed. This may allow us to identify pathophysiologic mechanisms linked to defects in mitochondrial dynamics and to develop therapeutic approaches.



 XML Download
XML Download