

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
2,3,7,8-Tetrachlorodibenzo-p-dioxin (2,3,7,8-TCDD) is a persistent environmental toxicant in the dioxin family of toxic chemicals and is structurally related to polyhalogenated aromatic hydrocarbons [1]. Humans are exposed to 2,3,7,8-TCDD in food, drinking water, soil, smoke, and air. Persistent exposure to 2,3,7,8-TCDD induces reproductive toxicity, such as reduction in female reproduction, and decreased sperm counts in male rodents [2,3]. Additionally, 2,3,7,8-TCDD suppresses immune response in laboratory animals, but promotes inflammatory responses by up-regulating pro-inflammatory cytokines such as interleukin-1 (IL-1) and tumor necrosis factor-alpha (TNF-α) [4]. Furthermore, 2,3,7,8-TCDD is carcinogenic to humans and rodents by affecting cellular necrosis, cell proliferation, and promoting tumor production [5]. In particular, 2,3,7,8-TCDD induces hepatotoxicity including hepatocellular swelling, degradation, and necrosis [6]. Liver damage induced by 2,3,7,8-TCDD is related to oxidative damage such as enhanced lipid peroxidation and decreased glutathione [7,8], and inflammatory cell infilteration by inducing cytochrome P450 genes through activation of the aryl hydrocarbon receptor (AHR) and its dimerization with AHR nuclear translocator [9,10].
Heat shock proteins (Hsps) are important chaperone proteins produced endogenously. Hsps are named according to their molecular weight, including Hsp90, Hsp70, Hsp60, and small heat-shock proteins (sHsps, <30 kDa). Hsps are induced in response to a variety of stressors including elevated temperature, hypoxia, inflammation, and ischemia [11]. They function to assist protein folding and conformation, and prevent undesirable protein aggregation. Additionally, Hsps are triggered as a protective mechanism against adverse stimuli as part of the stress response [12]. For example, dioxins induce Hsp70expression to reduce the toxicity of dioxin as a self-defense system [13]. 2,3,7,8-TCDD increases a sHsp, αB-crystalline gene in the eyes of experimental animals [14].
Oxidative stress is caused by increased production of reactive oxygen species (ROS) [15], which increases the risk for chronic diseases such as diabetes, cancers, neurodegenerative disease and liver cirrhosis [16]. To protect cells against oxidative stress, living organisms have developed an antioxidant defense system including catalase, superoxide dismutase (SOD), glutathione synthase (GST), and glutathione peroxidase (GPX) [17].
Both Hsp and antioxidant enzyme expression increases in response to stimuli or toxic chemicals. However, how these two classes of proteins are related in terms of cellular protection is relatively unknown. Therefore, we examined the effect of 2,3,7,8-TCDD on Hsp and antioxidant enzyme gene expression by real-time RT-PCR to clarify how 2,3,7,8-TCDD-induced hepatotoxicity affects Hsps and antioxidant enzymes.
METHODS
Experimental animals
Male Sprague Dawley rats (weight 173±5 g) were obtained from Charles River Laboratories, Inc. (Wilmington, MA, USA). They were housed under standard laboratory conditions (24±2℃; humidity, 50±10%, and 12-h day/night cycles). Animals were allowed to acclimate to the facility for 1 week and were provided standard chow diet and drinking water ad libitum. Animals were 6-weeks old on the first day of the exposure study.
Experimental design and tissue preparation
Sixty rats (20 per group) were divided into three groups: 1) control group (vehicle only), 2) 30 µg/kg 2,3,7,8-TCDD exposure group, and 3) 60 µg/kg 2,3,7,8-TCDD exposure group [18,19]. 2,3,7,8-TCDD (Cerilliant CIL Inc., Austin, TX, USA) was dissolved in corn oil and injected intraperitoneally, whereas corn oil alone was injected intraperitoneally into rats in the control group. To evaluate the acute toxic effect of 2,3,7,8-TCDD, five rats per group were sacrificed 1, 2, 4, or 8 days after the 2,3,7,8-TCDD injection. All animal experiments were performed according to the guidelines of the Animal Care Institutional Review Board of Korea University. After sacrifice under 70% CO2, rat livers were perfused with 50 ml cold PBS to eliminate excess blood, collected, immediately frozen in liquid nitrogen, and stored at -70℃ for later analysis.
RNA isolation
Total RNA was isolated from rat liver tissues using TRIzol reagent (Invitrogen Corp., Carlsbad, CA, USA), following the manufacturer's protocol. RNA integrity was assessed using an Agilent 2100 Bioanalyzer (Agilent Technologies, Palo Alto, CA, USA) and RNA was stored at -70℃ until further processing.
Real-time RT-PCR
Real-time PCR was performed to analyze the differential expression of Hsp genes using a Roche LightCycler 480 System (Roche Diagnostics, GmbH Mannheim, Germany) and the Taqman method using the Roche Universal ProbeLibrary (UPL) kit. Relative gene expression was determined by the comparative CT method, which is defined as the number of cycles required for the fluorescent signal to cross the threshold. All reactions were conducted in a total reaction mixture volume of 20µl containing 10.0µl 2× UPL Master Mix, 1.0µl 5' primer (10 pmol/µl), 1.0µl 3' primer (10 pmol/ml), 0.2 µl UPL probe, 1.0µl cDNA, and 6.8µl sterile water. The thermal cycling conditions for PCR were an initial denaturation for 10 min at 95℃, followed by 40 cycles of 60℃ for 10s, and 72℃ for 30s. Primers were designed with the Roche ProbeFinder assay tool (Table 1). Negative controls (except templates) were included in the PCR reaction to ensure specific amplification. LightCycler 480 software version 1.2 (Roche) was used to analyze the quantitative PCR. Values obtained from each sample were normalized to glucose-6-phosphate dehydrogenase expression.
RESULTS
Effect of 2,3,7,8-TCDD on the induction of sHsps
We evaluated gene expression changes in sHsps, Hsp27, Hsp10, and α-B-crystallin in livers of 2,3,7,8-TCDD-exposed rats (Fig. 1). Injecting 30 µg/kg 2,3,7,8-TCDD slightly but insignificantly increased Hsp27 expression 1 day after the injection. The level of Hsp27 was significantly increased following 60 µg/kg 2,3,7,8-TCDD injection, and Hsp27 mRNA levels were elevated over two-folds. In contrast, Hsp27 level dropped to control levels on day 2 and was down-regulated on day 4 compared to that in controls, even though it was not statistically significant in rats exposed to 30 and 60 µg/kg 2,3,7,8-TCDD. Hsp27 levels returned close to control levels after 8 days of exposure.
Hsp10, also known as chaperonin 10, was evaluated by real-time PCR. Rats exposed to 30 or 60 µg/kg 2,3,7,8-TCDD exhibited the slight but insignificant increase of Hsp10 expression levels on day 1 compared to those in the controls. Hsp10 levels was close to the controls after 8 days of exposure.
α-B-Crystallin levels were up-regulated 1 day after 30 and 60 µg/kg 2,3,7,8-TCDD exposure, but the up-regulation was only significant with exposure to 60 µg/kg. In contrast, α-B-crystallin levels 2 days after injection decreased dramatically compared to those after 1 day exposure, which was even significantly lower than that of the controls. α-B-crystallin levels were the lowest at 2 days, but gradually returned to control levels after 8 days of exposure.
Effect of 2,3,7,8-TCDD on Hsp70s
Hsp70 levels were slightly but insignificantly elevated after 1 day exposure to 30 or 60 µg/kg 2,3,7,8-TCDD compared to the controls, but decreased at 2 days after both concentrations of 2,3,7,8-TCDD exposure and did not change up to 8 days after 2,3,7,8-TCDD injection (Fig. 2A).
The levels of mortalin, also known as GRP75, were also determined by real-time PCR. Exposure to 60 µg/kg 2,3,7,8-TCDD significantly increased mortalin expression after 1 day. In contrast, mortalin levels dropped close to the controls after 2 days of injection and then signifcanlty decreased after 4 days compared to the controls, but recovered to control levels by 8 days in 60 µg/kg 2,3,7,8-TCDD treatment groups (Fig. 2B). However, 30 µg/kg 2,3,7,8-TCDD did not have significant effect on the expression of mortalin compared to the controls during the experimental period.
Effect of 2,3,7,8-TCDD on Hsp90s
Hsp90α levels were determined by real-time PCR. As shown in Fig. 2C, Hsp90α increased significantly by 2.5-fold after 1 day compared to the controls, but then decreased significantly at 2 days in rats exposed to both 30 and 60 mg/kg 2,3,7,8-TCDD, compared to 1 day. Hsp90α levels recovered to those of the control after 8 days. Similarly, Hsp90β increased up to 2.5-fold after a 1 day exposure to 30 and 60 µg/kg 2,3,7,8-TCDD compared to the controls. These increases decreased significantly after 2 days compared to 1 day and then recovered to control levels by 8 days (Fig. 2D). All Hsp90s showed a similar expression pattern by following 2,3,7,8-TCDD exposure.
Effect of 2,3,7,8-TCDD on other Hsps
The effects of 2,3,7,8-TCDD on glucose-regulated protein (GRP78) expression were evaluated by real-time PCR (Fig. 3A). GRP78, also known as Hspα5, was up-regulated in rats after a 1 day exposure to. GRP78 levels in rats exposed to 30 or 60 µg/kg of 2,3,7,8-TCDD for 1 day were not different from each other. After 2 days of 2,3,7,8-TCDD exposure, GRP78 levels decreased dramatically to levels lower than the controls in case of 60 µg/kg 2,3,7,8-TCDD injection. The decreased GRP78 levels recovered close to control levels after 8 days.
A 30 or 60 µg/kg 2,3,7,8-TCDD injection increased Hsp105 expression by 3.6 and 4.2-fold, respectively, in 1 day, but they were not significantly different from each other. Hsp105 levels dramatically decreased close to the control 2 days after the 2,3,7,8-TCDD injection (Fig. 3B).
Exposing rats to 30 or 60 µg/kg 2,3,7,8-TCDD increased calreticulin expression after 1 day, but the levels were decreased after 2 days, but those levels were not statistically significant compared to the controls (Fig. 3C).
Effect of 2,3,7,8-TCDD on antioxidant enzymes
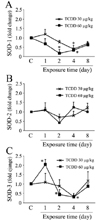
The effect of 2,3,7,8-TCDD on antioxidant enzyme mRNA expression was determined by real-time PCR. SOD-1, -2, and 3 expressions were determined separately with specific primers (Fig. 4). 2,3,7,8-TCDD slightly increase SOD-1expression after 1 day of exposure to 30 µg/kg compared to that in the control, but the increase was not significant. 2,3,7,8-TCDD exposure for 4 days significantly decreased SOD-1 levels, which recovered to control levels after 8 days in case of 30 µg/kg 2,3,7,8-TCDD. SOD-1 levels were significantly lower than those in the control at 2 and 4 days after injection of 60 µg/kg 2,3,7,8-TCDD and returned to control levels in 8 days. However, SOD-2 levels of rats exposed to 30 or 60 µg/kg 2,3,7,8-TCDD were not significantly different from the controls regardless of the exposure time in rats. In contrast, 60 µg/kg of 2,3,7,8-TCDD significantly increased SOD-3 expression at 1 day, the levels dropped lower than those of the control at 2 and 4 days, but recovered to control levels after 8 days. On the other hand, 30 µg/kg of 2,3,7,8-TCDD didn't exhibited significant effect on the expression of SOD-3 in 1, 2 and 8 days compared to the controls, but the levels of SOD-3 was significantly lower than the controls only in 4 days of injection.
The effect of 2,3,7,8-TCDD on catalase was evaluated by real-time PCR (Fig. 5A). Exposure of rats to 30 µg/kg 2,3,7,8-TCDD significantly decreased catalase expression at 4 days and the level was returned to the control levels at 8 days after exposure. Meanwhile, 60 µg/kg 2,3,7,8-TCDD significantly increased catalase levels compared to those in the control at 1 day after exposure. Conversely, exposure to 2,3,7,8-TCDD dramatically decreased catalase levels even lower than the controls on day 4. The decreased catalase levels following exposure to 60 µg/kg 2,3,7,8-TCDD did not recover to control levels after 8 days of exposure.
2,3,7,8-TCDD (60 µg/kg) exposure significantly increased GST levels by over two-fold in 1 day (Fig. 5B). The increased GST levels following 2,3,7,8-TCDD exposure were significantly lower than the control levels after 2 days of exposure, and recovered to the control levels after 4 days. Additionally, GPXs, GPX-1 and GPX-4 mRNA levels were also evaluated by real-time PCR in livers from 2,3,7,8-TCDD-exposed rats (Fig. 5C, D). Exposure to 2,3,7,8-TCDD for 1 day didn't have significant effect on the levels of both GPXs, but GPX levels dropped dramatically at day 2 in the 60 µg/kg 2,3,7,8-TCDD-exposed rats and slowly recovered to the control levels. However, in case of 30 µg/kg 2,3,7,8-TCDD exposure, GPX levels were only significantly decreased at 2 days after exposure than the controls, and those levels were returned close to the control after 4 days.
DISCUSSION
We studied the effects of 2,3,7,8-TCDD on Hsps and antioxidant enzymes expression. The livers from rats exposed to 30 or 60 µg/kg 2,3,7,8-TCDD were evaluated for mRNA expression of Hsps and oxidative markers (antioxidant enzymes). Our results indicated that 2,3,7,8-TCDD increased the levels of some Hsps and antioxidant enzymes in 1 day, dramatically decreased the levels to lower than those in the controls by 2-4 days, and then the levels returned to those of the control after 8 days of exposure.
The heat shock response is a dramatic up-regulation of Hsps by the stimuli such as increased temperature or other stressors. It is induced as a protective mechanism against the non-specific toxicity produced by generating abnormal proteins and altered cellular functions [12]. Therefore, the stress induced by 2,3,7,8-TCDD exposure triggered an increase in Hsp expression, and allowed the animal model to withstand the toxicity induced by 2,3,7,8-TCDD. Additionally, prolonged toxicity by 2,3,7,8-TCDD depleted Hsp levels, but recovered to normal levels following a reduction in 2,3,7,8-TCDD toxicity. Another possibility for the decrease in Hsp expression could be related to threshold limits in the cell [20,21].
Hsp70 is a chaperone that functions in protein folding and protection of cells from stress and is one of the most studied Hsps. Hsp70 overexpression in early hepatocellular carcinoma has been previously reported [22,23]. Additionally, some environmental toxicants have been reported to induce Hsp70. For example, exposure of Drosophila melanogaster to benzene, toluene, or xylene induces Hsp expression including Hsp70 [24]. Mercury, cadmium, and arsenite induce Hsp70 [25]. 2,3,7,8-TCDD exposure also significantly increases Hsp70 mRNA expression in livers of rats and marmosets [13,26]. Furthermore, Hsp70 overexpression protects mouse embryos against the teratogenic effects of arsenite [27]. Induction of Hsp70 family proteins by dioxin has been associated with a reduction in its toxicity [13]. In particular, induction of Hsp70 is correlated with an anti-apoptotic effect in the liver by interfering with caspase-dependent apoptotic pathways [28]. In our study, exposure of rats to 2,3,7,8-TCDD significantly increased Hsp70 mRNA expression in livers after 1 day of exposure. Therefore, Hsp up-regulation in the liver after short exposure to 2,3,7,8-TCDD could be explained due to protective mechanisms against 2,3,7,8-TCDD-induced toxicity.
In accordance with Hsp70, other Hsps including GRPs, Hsp90s, and sHsps elevated increased following exposure to 2,3,7,8-TCDD. Hsp90 is a chaperone that functions as a ligand for binding of the aryl hydrocarbon receptor (AHR) [29]. Exposure of striped bass to copper increases Hsp90 expression in liver tissues [30]. GRP78 is over-expressed in cultured human cells by volatile organic compounds such as benzene, ethylbenzene, toluene, xylene, and chlorinated derivatives [31]. In addition, Hsp24 and Hsp90 levels are enhanced by exposure of chick embryos to metals such as mercury, cadmium, and arsenite [25]. Mouse embryonic stem cells overexpress Hsp27 following exposure to cadmium, mercury, and arsenite to protect themselves against death [32]. These reports suggest that increased Hsp expression following metal exposure might be correlated with the protective mechanisms of organisms against developmental toxicity.
Previous studies have suggested that oxidative stress following ROS production plays a role in 2,3,7,8-TCDD-induced toxicity [7], whereas others have observed superoxide production, lipid peroxidation, and oxidative damage in fetal and placental tissues upon treatment with TCDD [33,34]. Cyp1A1 overexpression contributes to ROS generation and subsequent oxidative stress in mammalian cells [35]. 2,3,7,8-TCDD administration also induces hepatic lipid peroxidation (as determined by TBARS) by as much as 7-fold in rats [36]. These studies imply that 2,3,7,8-TCDD-induced oxidative stress in our study.
Up-regulation of oxidative stress contributed to the toxic effects of 2,3,7,8-TCDD. ROS increases in the testis of 2,3,7,8-TCDD-injected mice [37], which is dependent on an increase in mitochondrial respiration [38]. 2,3,7,8-TCDD also increases catalase and GPX production in the brains of exposed rats [39]. Moreover, treatment of adipocytes with 2,3,7,8-TCDD increases SOD and catalase activity [40]. 2,3,7,8-TCDD also increases the cytotoxicity of rat Sertoli cells, which is accompanied by oxidative stress including SOD, catalase and GPX up-regulation [41].
ROS induces Hsps [42,43]. Hsp90 overexpression induced by NaHS in PC12 cells is accompanied by increased ROS production [44]. In addition, chromium-treated murine embryonic liver cells show increased production of ROS, and this is accompanied by elevated Hsp expression such as Hsp27 and Hsp70 [45]. Furthermore, human airway epithelial cells exposed to nickel increase oxidative stress associated with ROS, and up-regulate Hsp70 [46]. In our study, exposure of rats to 2,3,7,8-TCDD induced not only Hsp production, but also antioxidants enzymes in a similar manner. These results suggest that 2,3,7,8-TCDD-induced oxidative stress could be attributed to up-regulation of Hsps in the rat livers.
2,3,7,8-TCDD is a environmental toxicans, which has been reported to induce diverse adverse effect on living organisms. In particular, 2,3,7,8-TCDD is well-known to induce hepatotoxicity by single or multiple administration with dose and time-dependent manner. A administration of 200 µg/kg 2,3,7,8-TCDD significantly increased the hepatocellular hypertrophy [47]. In addition, IP injection of 5 to 500 µg/kg 2,3,7,8-TCDD significantly increased the number of multinucleated and swelling hepatocytes in addition to the serum cholesterol [48]. Furthermore, 50 µg/kg 2,3,7,8-TCDD increased serum fatty levels with plasma membrane damage in the livers [49].
In conclusion, rats were intraperitoneally exposed to 2,3,7,8-TCDD and livers were dissected for real-time PCR to clarify the effect of 2,3,7,8-TCDD on Hsps and antioxidant enzymes. As a result, 2,3,7,8-TCDD exposure (60 µg/kg 2,3,7,8-TCDD) significantly increased Hsp expression such as Hsp27, α-B-crystallin, mortalin, Hsp105 and Hsp90s after a 1 day exposure, and 2,3,7,8-TCDD also increased SOD-3, GTS, and catalase expression after1 day of exposure. However, the some of Hsps (α-B-crystallin, Hsp90, GRP78) and antioxidant enzymes expression (SOD-1, SOD-3, catalase, GST, GPXs) were down-regulated on days 2 or 4, but recovered to control levels on day 8. These results suggest that Hsps and antioxidant enzymes helped the animals withstand 2,3,7,8-TCDD-induced hepatotoxicity, but were depleted by long exposure to 2,3,7,8-TCDD, and then recovered to normal levels through a reduction in 2,3,7,8-TCDD hepatotoxicity.





 XML Download
XML Download