

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
The progression of type 1 diabetes (T1D) starts with immunological misdirection. This pathogenic misdirection results in an inflammatory environment in the pancreatic islets and ultimately, the death of pancreatic β-cells. As these cells are the primary source of insulin in the body, glucose homeostasis is compromised, and T1D is manifested [1,2]. Numerous studies have reported the role of diabetogenic T cells in the progression of T1D, however the involvement of macrophages in the pathogenesis of this disease has been investigated as well [3]. In this study we explorered the role of macrophages in the pathogenesis of type 1 diabetes. The pancreatic microenvironment was simulated using RAW264.7 cells as macrophages, and MIN6 cells as pancreatic β cells. The anti-oxidant enzyme peroxiredoxin-1 (PRX-1) was knocked down in both cell types to observe how the presence or the absence of this enzyme affects this system.
RAW264.7 cells are a well-established cell line for macrophage studies. Macrophages can be described as one most active secretory cell types in the body, releasing a wide range of mediators that regulate inflammation, adaptive immunity, and homeostasis. Macrophage activation, caused by infection and other immunological stimuli, causes overproduction of inflammatory mediators including nitric oxide (NO) and pro-inflammatory cytokines such as the tumor necrosis factor-α (TNF-α) and interleukin-1β (IL-1β), IL-6, and IL-12 [4]. RAW264.7 cells were stimulated with lipopolysaccharide (LPS) to prime them for active production of inflammatory mediators.
The MIN6 mouse insulinoma cell line is morphologically similar to pancreatic β cells, and exhibits glucose stimulated insulin secretion [5]. This cell line was co-cultured with RAW264.7 cells, which were stimulated with lipopolysaccharide to prime them for active production of inflammatory mediators.
Peroxiredoxin (PRX) is an anti-oxidant enzyme in living organisms, scavenging H2O2 and alkyl peroxides [6]. The expression of PRX-1 and PRX-2 is increased in pancreatic β cells when stimulated with inflammatory cytokines, suggesting an involvement of this enzyme in the pathogenesis of T1D [7]. Our previous studies were focused on the effects of PRX-1 knockdown in RAW264.7 cells [8]. In this current study, PRX-1 was knocked down using a lentiviral system in both MIN6 and RAW264.7 cells to observe the effects of PRX-1 knockdown under co-culture conditions.
Go to : 
METHODS
Cell culture
MIN6 cells were obtained from the Korean Cell Line Bank, RAW264.7 macrophage cell lines were obtained from the American Type Culture Collection (Manassas, USA) and both were maintained in DMEM medium (GIBCO) supplemented with 10% heat inactivated FBS (fetal bovine serum, CELLGRO), 2 mM L-glutamine, 100 U/ml penicillin and streptomycin (CELLGRO) at 37℃ in a 5% CO2 humidified incubator. Cells were cultured in 25T flasks (SPL). Co-culture of MIN6 and RAW264.7 cells were conducted using a 0.4 µM cell culture insert (Becton Dickinson).
Reagents
Capture and detection antibodies used in ELISA for TNF-α and IL-6, IL-10 was purchased from BD Biosciences (San Jose, CA, USA) and antibodies for IL-1β were purchased from eBioscience. Polybrene for transduction was obtained from Macrogen. The amount of generated hydrogen peroxide in the supernatant was detected using Amplex® Red Hydrogen Peroxide/Peroxidase Assay Kit (invitrogen, Carlsbad). Capture and detection antibodies used in ELISA for TNF-α and IL-6 was purchased from BD Biosciences. Polybrene for transduction was obtained from Macrogen.
Establishment of the PRX-1 knockdown cell line
The complete sequence for PRX-1 (Gene ID: NM_011034) was found by using PubMed. Four target sequence candidates of PRX-1 for gene silencing were determined. Selected candidate sequences are as following: candidate 1 (TRCN 0000120688, Sigma-Aldrich, St. Louis), 5'-gcttcagtgatagagc cgat-3'; candidate 2 (TRCN0000120690, Sigma-Aldrich, St. Louis), 5'-cagtgatagagccgat-3'; candidate 3 (sequence determination was made through the use a program provided by www.siRNAwizard.com), 5'-gtgatagagccgatgaattta-3'; candidate 4 (sequence determination was made through the use a program provided by www.siRNAwizard.com), 5'-gatg tatatgtgaggctagta-3'.
The shRNA lentiviral vector targeting peroxiredoxin-1 (shPRX-1) was established using synthetic double strand oligonucleotides (Macrogen, Seoul). The construct was designed to produce shRNAs promoted from the U6 promoter and to express GFP from the hCMV promoter. This shPRX-1 construct was used to generate stable transduced RAW264.7 and MIN6 cells. A scramble (Macrogen, Seoul) was used as a negative control. To silence PRX-1 in RAW264.7 and MIN6 cells, 1×106 cells were seeded per well in a 6-well plate and were cultured overnight. After removal of the media, viral particle soup containing the vector and 6 µg/ml of polybrene (Macrogen, Seoul) made to a final volume of 1 ml was added to the 6-well plate. After an overnight incubation the media was replaced with fresh media. An enriched population of RAW264.7 and MIN6 cells transduced with a green fluorescent protein (GFP) construct were incubated in 10% fetal bovine serum (FBS) Dulbecco's modified eagle media (DMEM) containing puromycin (4 µg/ml) for selection and fluorescence-activated cell sorting (FACS).
Cell viability assay
Cells were seeded at 3×104 cells per well and pre-cultured in the 96-well plates before for 12 hr. Afterwards, a cytokine mix (IL-1β, TNF-α, IFN-γ each 5 ng/ml) was treated with media for 24 hr. Subsequently, 10 µl of MTT [3-(4,5-dimethylthiazolyl-2)-2, 5-diphenyltetrazolium bromide] solution (5 mg/ml, Sigma-Aldrich, St. Louis) was added to each well and incubated for 2 hr. After incubation, 100 µl of solubilization solution (0.04 N HCL in isopropanol) were added to each well and the plate was evaluated at 570 nm wavelength using a microplate reader (Emax, Molecular Devices).
NO assay
Supernatants from cell cultures were assessed for nitric oxide production. An equal volume of Griess reagent (Sigma, St. Louis) was added to the supernatant from each well. The absorbance of the mixture at 540 nm was determined using a microplate reader (Emax, Molecular devices), and nitrite concentration was determined using a dilution of standard nitrite as control.
Total RNA isolation, reverse transcription and polymerase chain reaction (RT-PCR)
Total RNA was isolated from each sample using Trizol reagent (Invitrogen, Carlsbad, California, USA) in according to the manufacturer's instructions. RNA was transcribed at 42℃ for 1 hr in a volume of 25 µl containing 5× RT buffer, 10 mM dNTPs 200 units Maloney murine leukemia virus (M-MLV) reverse transcriptase (Promega, Madison, Wisconsin, USA) and 100 pmole of oligo-dT primer. cDNA from each sample was amplified with 10 pmole of each oligonucleotide primers, rTaq Plus 5× PCR master mix (ELPIS biotech, Daejeon, Chungcheongnam-do, South Korea) in a final volume of 20 µl. PCR was performed after quantitative normalization for each gene by densitometry using β-actin gene expression. Then, cDNA was amplified by PCR using primers specific for the mouse Bcl-xL, Bim, iNOS, and β-actin. Densitometry analysis was performed using Image J software (Wayne Rasband National Institutes of Health, USA, Ver 1.45s).
Enzyme-linked immunosorbent assay (ELISA)
96-well plates were coated overnight at 4℃ with the capture antibody. Each well was washed three times with PBS-T, incubated for 1 hr with blocking solution at room temperature, and then washed four times with PBS-T. Samples and diluted standards were added to the plate and incubated overnight at 4℃. Following 4 wash cycles, the detection antibody was added. After 45 min incubation at room temperature, the wells were washed and avidin-conjugated alkaline phosphatase was added at room temperature and incubated for 30 minutes. Subsequently, the substrate solution was added and the plates were maintained at room temperature for 5 to 30 minutes before the addition of the stop buffer. The absorbance was read at 405 nm using a microplate reader (Emax, Molecular Devices).
Fluorescence microscopy
1×106 MIN6 cells were seeded on a 60Φ dish and cultured for 12 hr. After culture, the media was discarded and cells were prepared and observed using a Leica DM2500 microscope. Image capture was performed using the Leica Qwin software.
Flow cytometry
shPRX-1 vectors that express eGFP and PRX-1 sense, anti-sense sequence were transduced to cells using polybrene. Transduced cells were cultured in 10% FBS DMEM containing puromycin (2 µg/ml) for selection. After selection, cells were harvested and 1×106 cells were added to per FACS tube. Fluorescence acquisition and analysis were performed using FACScan and Cellquest Pro (BD biosciences, San Jose).
Western blot analysis
Whole cell proteins were extracted in RIPA buffer (Elpis Biotech, Daejeon, South Korea). The extracts were separated by SDS-PAGE and transferred to a PVDF membrane. Blots were blocked and incubated with anti-PRX-1 (Ab frontier), or anti-β-actin antibodies (Cell Signaling, Danvers), or anti-STAT3, anti-phospho-STAT3 antibodies at 4℃ overnight. The membranes were then incubated with anti-mouse IgG or anti-rabbit IgG HRP-linked antibodies (Cell Signaling), and developed using the ChemiGlow West (Alpha Innotech).
Statistical analysis
Data are expressed as mean±SD, and statistical analysis were performed by using a Student's t test. p values of <0.05 were considered statistically significant.
Go to :
RESULTS
Knockdown of PRX-1 through PRX-1 shLenti constructs in MIN6 and RAW264.7 cells
Four candidates were considered as targets for knockdown, and candidate two became the final target sequence because it exhibited the most overlap (Fig. 1). The method of knockdown of PRX-1 in RAW264.7 cells has been described in our previous work [8]. In order to silence PRX-1 in MIN6 cells, 1×106 cells were seeded per well in a 6-well plate and cultured overnight, viral soup containing the viral vector and polybrene was added subsequently. To establish transduced MIN6 cells, scramble (empty vector control) and shPRX-1 construct were transduced into MIN6 cells. For selection of transduced cells, cells were incubated with puromycin for two weeks. Transduction was confirmed through fluorescence microscopy and FACS (Fig. 2A). RT-PCR and western blot techniques were employed to confirm knockdown of PRX-1 at protein level and gene level (Fig. 2B). When compared with scramble infected cells, shPRX-1 infected cells showed 80% decrease in the expression of PRX-1. Thus, the knockdown of PRX-1 by the shPRX-1 construct was confirmed.
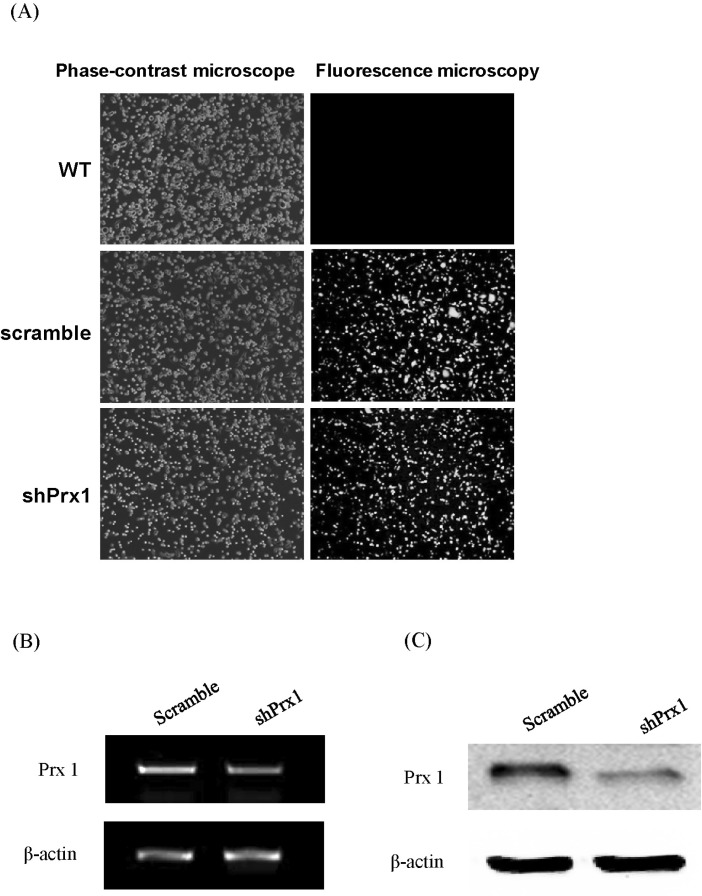 | Fig. 2(A) shPRX-1 construct and empty vector transduction efficiency. WT, scramble, and shPRX-1 1×106 MIN6 or RAW264.7 cells were harvested in PBS 1 ml. Then, the transduction efficiency was detected by phase fluorescence microscopy, representative imagery are shown. WT: wild type; Scramble: empty vector; shPRX-1: PRX-1-knockdown. (B) shPRX-1 silenced PRX-1 at gene level. Scramble, and shPRX-1 2×105 MIN6 or RAW264.7 cells were incubated in 24 well plate overnight. Total mRNA were extracted by Trizol and verified by RT-PCR, representative bands are shown. (C) shPRX-1 silenced PRX-1 at protein levels. Scramble, and shPRX-1 2×106 MIN6 or RAW264.7 cells were incubated in 6 well plate overnight. Whole protein was extracted by RIPA buffer and verified by western blot analysis, representative blots are shown.
|
Determination of optimal condition of RAW264.7 cells for co-culture
In this simulated microenvironment, RAW264.7 cells played the part as the messengers of inflammation and MIN6 cells the victim. First, RAW264.7 cells were stimulated with LPS at varying time intervals from 0 hr to 24 hr to determine when they produce the largest amount of inflammatory mediators. TNF-α and nitric oxide (NO) levels were determined at each time interval. TNF-α production held steady until 4 hr and dropped precipitately afterwards (Fig. 3A). On the other hand, NO production increases proportionally with time until 12 hr, when a decrease is observed in NO production levels (Fig. 3B). To observe the effects of PRX-1 knockdown in the simulated T1D microenvironment, 4 hr LPS stimulation of RAW264.7 cells prior to co-culture with MIN6 cells was selected as the optimal culture condition.
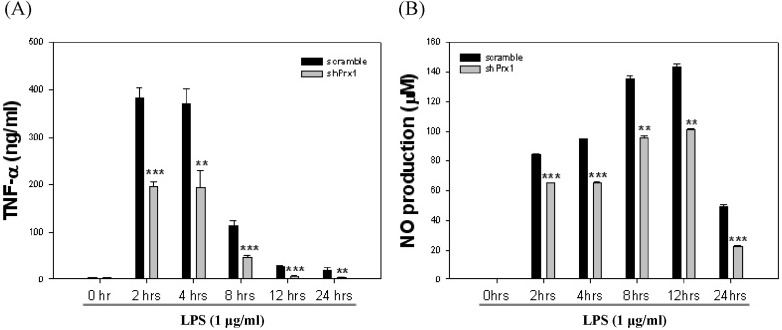 | Fig. 3Determination of optimal stimulation of RAW264.7 cells for co-culture. (A) RAW264.7 cells were stimulated with LPS (1 µg/ml) at varying time intervals from 0 hr to 24 hr, TNF-α levels were evaluated by ELISA at each time interval. Values represent the mean±SD (**, p<0.01 vs. scramble, ***, p<0.005 vs. scramble). (B) RAW264.7 cells were stimulated with LPS (1 µg/ml) at varying time intervals from 0 hr to 24 hr, nitric oxide (NO) levels were determined by NO assay at each time interval. Values represent the mean±SD (**, p<0.01 vs. scramble, ***, p<0.005 vs. scramble).
|
MIN6 cells co-cultured with LPS-stimulated shPRX-1 RAW264.7 cells in the presence of L-NMMA showed increased anti-apoptotic gene expression compared to MIN6 cells co-cultured with RAW264.7 cells
RAW264.7 cells were stimulated with LPS for 4 hr in this study before being co-cultured with MIN6 cells. When scramble MIN6 cells were co-cultured with shPRX-1 RAW264.7 cells, scramble MIN6 cells displayed increased iNOS expression and decreased expression of an established pro-apoptotic gene, Bim (Fig. 4A). In order to clear up the picture and observe the effects of cytokines in this system, L-NMMA, an iNOS inhibitor was treated to RAW264.7 cells to exclude the effects of NO on MIN-6 cells. Interestingly, after the effect of NO was removed, scramble MIN6 cells co-cultured with shPRX-1 cells showed increased expression of Bcl-xL, an anti-apoptotic gene. Contrastingly, when shPRX-1 MIN6 cells are coupled with scramble RAW264.7cells, a significant increase in the expression of the pro-apoptotic gene Bim was observed (Fig. 4B). These results suggest that other mediators, other than NO, produced by RAW264.7 cells may influence the viability of MIN6 cells and that these mediators are affected by the expression of PRX-1.
 | Fig. 4Apoptotic gene expression in RAW264.7 and MIN6 co-cultures visualized through RT-PCR. (A) RAW264.7 cells were pre-stimulated with LPS for 4 hr before 24 hr of co-culture. Total RNA from each group was extracted and reverse transcribed for RT-PCR analysis. (B) Under the same conditions, L-NMMA was added to exclude the effect of NO and gene expression was assessed by RT-PCR.
|
Decreased NO production is observed in MIN6 cells when treated with IL-6 and PRX-1 is required in the cell survival signals induced by IL-6
MIN6 cells co-cultured with RAW264.7 cells were treated with IL-6 and IL-10 to determine whether NO production is affected by these cytokines. IL-6 is generally perceived as an inflammatory cytokine while IL-10 has been described as an anti-inflammatory cytokine [9]. Scramble and shPRX-1 MIN6 cells were treated with a mix of inflammatory cytokines (IL-1β, TNF-α, IFN-γ each 5 ng/ml) to simulate apoptotic conditions. Afterwards, they were treated with IL-6, IL-10 or a combination of both. The group treated with IL-6 exhibited significantly lower levels of NO production and slightly better viability (Fig. 5). While the group treated with only IL-10 did not exhibit significant differences in NO production or cell viability (Fig. 5).
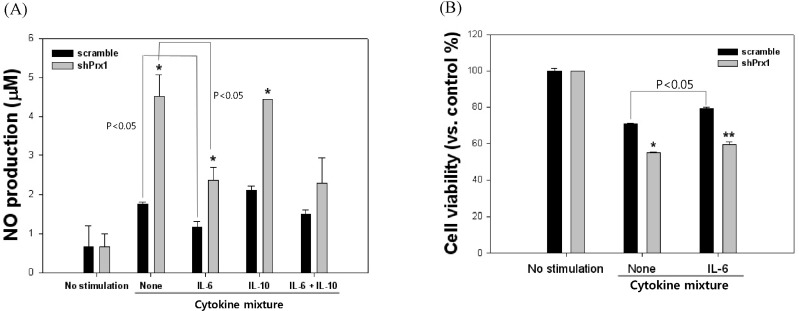 | Fig. 5NO production and cell viability of scramble and shPRX-1 MIN6 cells in the presence or absence of IL-6 and IL-10. (A) NO production of MIN6 cells were observed after treatment with an inflammatory cytokine mixture for 24 hr and the addition of IL-6, IL-10 or a combination of both. Values represent the mean±SD (*, p<0.05 vs. scramble, **, p<0.01 vs. scramble). (B) Cell viability of MIN6 cells were analyzed after treatment with an inflammatory cytokine mixture for 24 hr and in the presence or absence of IL-6. Values represent the mean±SD (*, p<0.05 vs. scramble, **, p<0.01 vs. scramble).
|
These results signify that IL-6 has a significant protective effect in MIN6 cells under apoptotic conditions and shPRX-1 cells showed decreased viability compared to scramble cells under the same conditions. To ascertain the cause of this increased survivability of MIN6 cells, pro- and anti-apoptotic gene expression, and the expression of iNOS was visualized through RT-PCR (Fig. 6). In the presence of inflammatory cytokines, scramble MIN6 cells treated with IL-6 exhibited escalated expression levels of the anti-apoptosis gene Bcl-xL while shPRX-1 MIN6 cells displayed decreased expression of this molecule. The contrary was observed for the pro-apoptosis gene Bim, with shPRX-1 MIN6 cells showing higher levels of Bim expression. The expression of iNOS was significantly decreased with IL-6 treatment, however in shPRX-1 MIN6 cells, the decrease was less pronounced compared to scramble cells.
 | Fig. 6The expression of iNOS, Bcl-xL and Bim was assessed by RT-PCR. Under cytokine mix treatment, IL-6 down-regulates iNOS expression in a significant manner in scramble MIN6 cells compared to shPRX-1 cells. shPRX-1 MIN6 cells treated with IL-6 showed increased expression of the anti-apoptotic gene, Bcl-xL and decreased expression of the pro-apoptotic gene, Bax.
|
PRX-1 affects survival signals induced by IL-6 and also affects insulin production in MIN6 cells
As the rescue effect of IL-6 seemed to differ between scramble and shPRX-1 MIN6 cells (Fig. 5B), STAT3 phosphorylation levels were visualized through western blot in order to determine if the IL-6 signaling pathway was affected by knockdown of PRX-1. STAT3 is a well-established mediator of IL-6 signaling and translocates to the nucleus upon phosphorylation as homo- or heterodimers, where it acts as a transcriptional activator. It seems that while treatment with IL-6 increased STAT3 phosphorylation in MIN6 cells, shPRX-1 MIN6 cells showed diminished phosphorylation of STAT3 compared to control (Fig. 7). The data suggests that PRX-1 has a role in preserving IL-6 induced STAT3 activation in normal cells.
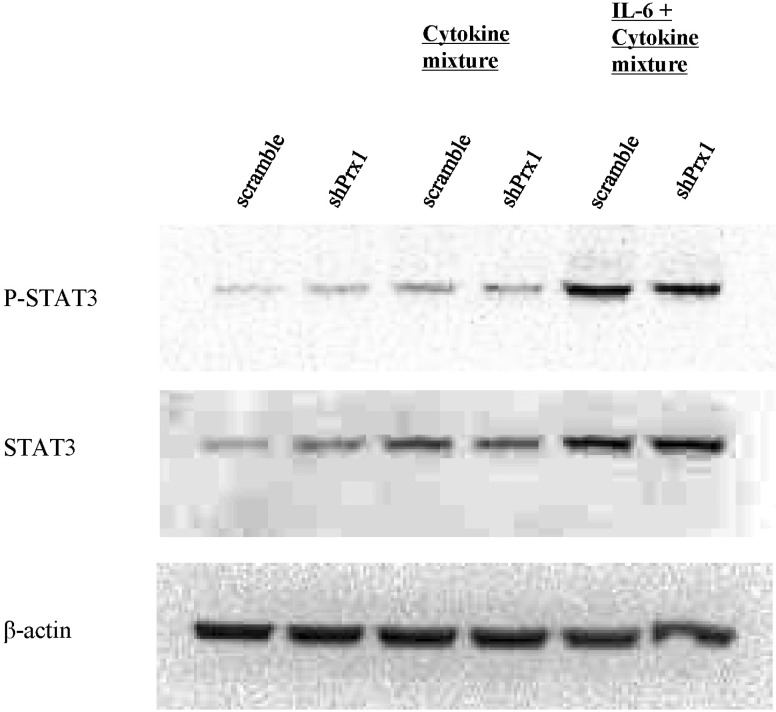 | Fig. 7PRX-1 promotes phosphorylation of STAT3. Whole protein was extracted through RIPA buffer and after performing SDS-PAGE electrophoresis, the protein was transferred to a PVDF membrane. The membrane was subsequently blocked and blots were visualized using antibodies for β-actin, phosphor-STAT3 and STAT3 and anti-mouse IgG or anti-rabbit IgG HRP-linked antibodies.
|
Go to :
DISCUSSION
In this study, RAW264.7 cells were co-cultured with MIN6 cells to simulate the T1D pathogenic microenvironment, with the anti-oxidative enzyme peroxiredoxin-1 knocked down in one or both cell types in order to elucidate the function of this enzyme in the pathogenesis of T1D. The protective effect of Bcl-xL, an anti-apoptotic gene in cytokine-induced β-cell apoptosis has previously been reported [10]. In our results, MIN6 cells co-cultured with LPS-stimulated shPRX-1 cells displayed increased expression of Bcl-xL as shown in Fig. 4B after L-NMMA was treated and it seems that the apoptosis-inducing ability of shPRX-1 RAW264.7 cells are somewhat inhibited compared to scramble RAW264.7 cells. While when shPRX-1 MIN6 cells are coupled with scramble RAW264.7 cells, the expression of the pro-apoptotic gene Bim was shown to be significantly increased (Fig. 4B). It seems that the knockdown of PRX-1 induces a certain vulnerability in MIN6 cells to RAW264.7 cell induced apoptosis. Supporting our findings, a recent study on peroxirodoxin-3, a family member of peroxiredoxin, showed that it protects pancreatic β-cells from apoptosis as well [11].
The protective effects of IL-6 on pancreatic β cells have been reported by numerous investigators [12-14]. As IL-6 is readily secreted by activated macrophages [15], it was hypothesized that IL-6 produced by RAW264.7 cells may be the cause of increased expression of Bcl-xL in co-cultured MIN6 cells. Also the anti-inflammatory cytokine, IL-10 has been reported to accelerate destruction of β-cells in nonobese diabetic mice [16]. However, in our results, while IL-6 decreased the production of NO in both scramble and shPRX-1 MIN6 cells under inflammatory cytokine stimulation, treatment IL-10 did not show any significant decrease in NO production (Fig. 5A). Also, IL-6 treatment increased the viability of MIN6 cells under inflammatory conditions (Fig. 5B). Interestingly, the protective effect of IL-6 was hindered in shPRX-1 MIN6 cells, suggesting a role for PRX-1 in preserving β-cell protective effects of IL-6. A similar trend was observed in gene expression levels of iNOS, Bcl-xL and Bim. The treatment with IL-6 increased the expression of the anti-apoptotic gene Bcl-xL, and decreased the expression of the pro-apoptotic gene Bim in scramble MIN6 cells (Fig. 6).
A recent study indicated that inhibition of the endogenous and exogenous IL-6 induced STAT-3 phosphorylation in human pancreatic cancer cells reduces cell viability [17]. Therefore, the phosphorylation of STAT3, which is the principal mediator of IL-6 signaling, was observed through western blot in this current study. This was performed in order to determine whether PRX-1 function affects the protective effects of IL-6. Compared to scramble cells, shPRX-1 MIN6 cells were shown to have lower levels of STAT3 phosphorylation in response to IL-6 (Fig. 7). The results suggest that PRX-1 deficiency results in downregulation of STAT3 phosphorylation, and may consequently hinder cytoprotective effects of IL-6 in pancreatic β cells.
Overall, our results suggest that the deficiency or loss of activity of PRX-1 in pancreatic β-cells will result in decreased viability of these cells and may expedite the progression of type 1 diabetes. Along with the previously reported cytoprotective effects of this antioxidant enzyme, it seems that PRX-1 affects the protective action of IL-6 on pancreatic β cells. This suggests new roles for PRX-1 in immunological dysregulation. Future studies utilizing in vivo models would shed light on the clinical implications of this enzyme and may open new doors in the treatment of type 1 diabetes.
Go to :
 XML Download
XML Download