

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
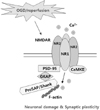
During brain ischemia, the lack of energy induces a collapse of ionic gradients, followed by excessive presynaptic neuronal depolarization, abnormal release of excitatory neurotransmitters, and a reduced reuptake of these neurotransmitters from the extracellular space. Glutamate is the major excitatory neurotransmitter in the brain, and an increased level of glutamate induces overstimulation of N-methyl-D-aspartate receptors (NMDARs) in the postsynapse, a massive influx of Ca2+, pathological synaptic plasticity, and the subsequent activation of cell death signaling pathways. This overstimulation of NMDARs is commonly referred to as excitotoxicity, and is assumed to be a critical mechanism in the neuronal death that occurs following an ischemic insult [1-4].
Most studies into ischemia have focused primarily on the mechanism of cell death or survival, while much less attention has been paid to the activity and morphology of synapses, which could have an important impact on cell function and survival [5-7].
Postsynaptic density (PSD) is a postsynaptic scaffold harboring several different receptor complexes, which directly participate in synaptic transmission [8-11]. In the mammalian CNS glutamatergic synapse, a typical PSD is composed of a large, complex protein network consisting of several hundred different proteins, in which members of the membrane-associated guanylate kinase (MAGUK) family are of crucial importance [12-15].
The PSD is composed of four major types of molecules: membrane-bound, cytoskeletal, and scaffolding proteins, as well as modulatory enzymes. The most abundant membrane-bound proteins found in the PSD are NMDARs, while the abundant cytoskeletal elements, including actin, are important in localizing and clustering the PSD receptors and signal complexes [16]. Scaffolding proteins, including PSD-95, bring the various PSD components into association. The key enzymes regulating PSD components include calcium/calmodulin protein kinase II (CaMKII), protein kinase C (PKC), and neuronal nitric oxide synthase (nNOS) [17].
A major component of the PSD fraction is PSD-95, which is the best characterized MAGUK protein. As an essential synaptic adaptor protein, PSD-95 interacts with a large variety of molecules, and thus by physically bringing together cytoplasmic signal transduction proteins and surface receptors, may facilitate the coupling of various signaling cascades within the PSD [11,18-20].
The first molecule identified as binding to the PDZ domain of PSD-95 was the NR2A/B subunit of the NMDAR [21-23]. NMDARs in the mammalian brain are thought to be heterotetramers of two obligatory NR1 subunits, in combination with either NR2A and/or NR2B [24-26].
CaMKII is one of the most abundant proteins in neurons, and is highly concentrated in the PSD at glutamatergic synapses [16,27,28]. It plays a key role in synaptic plasticity [29]. Two isoforms, α- and β-CaMKII, are neuron specific, and CaMKII shows dynamic translocation to the PSD in response to Ca2+ influx through NMDARs [30].
Therefore, this study aimed to investigate the effects of reperfusion following oxygen/glucose deprivation (OGD) not only upon neuronal cell death, but also on the ultrastructural and biochemical characteristics of PSD proteins in the stratum lucidum of the CA3 area in organotypic hippocampal slice cultures.
METHODS
Preparation of organotypic hippocampal slice cultures
All surgical and experimental procedures were reviewed and approved by the Institutional Animal Care and Use Committee (IACUC) of Ewha Womans University.
Organotypic hippocampal slice cultures were prepared from the hippocampi of 10-day-old Sprague-Dawley rats [31,32]. Hippocampi were dissected and cut at 300-µm thickness using a McIlwain tissue chopper (The Mickle Laboratory Engineering Corporation, UK). The slices were carefully placed onto membrane units (0.4 µm; Millicell-CM, Millipore, MA, USA) in 6-well plate, with each well containing 1.2 ml of a culture medium, as previously described [31], and were incubated at 37℃ in 95% O2/5% CO2.
Induction of OGD
Excitotoxicity and NMDARs contribute to cell death induced by OGD/reperfusion. To achieve the appropriate conditions for NMDAR expression [33], slices were grown in culture for 14 days. After incubation for 14 days, slices were washed with fresh media, and then placed in an OGD buffer pre-gassed with 95% N2/5% CO2, and then incubated at 37℃ in 95% N2/5% CO2 for 30 min. After this, the OGD media was replaced with normal, fresh media for up to 24 h during reperfusion. Control slices underwent the same manipulations, but were incubated with fresh normal media instead of OGD media gassed with 95% O2/5% CO2 at 37℃ for 30 min.
Transmission electron microscopy (TEM)
Slices were fixed in 2.5% glutaraldehyde at 4℃ for 2 h, postfixed in 1% OsO4 for 1 h, and embedded in an Epon 812. Before ultrathin cutting, the semi-thin sections (2 mm) were cut from the CA3 area of the slices, stained with toluidine blue, and examined under light microscopy. From each semi-thin section, 40~50 ultrathin sections (LKB Ultratome III, Mager Scientific, USA) were cut and stained with 5% uranyl acetate and lead citrate. The preparations were examined using a transmission electron microscope (Hitachi H-600, Hitachi, Tokyo, Japan).
For ethanolic phosphotungstic acid (EPTA) staining [34,35], sections were dehydrated in an ascending series of ethanol up to 100%, and stained for 45 min with 1% phosphotungstic acid (PTA), which was prepared by dissolving 0.1 g PTA in 10 ml of 100% ethanol, and adding 4 drops of 95% ethanol. Sections were then dehydrated in dry acetone, and embedded in Epon 812. Ultrathin sections (0.1 µm) were prepared and examined using an electron microscope (Hitachi H-600, Hitachi, Tokyo, Japan).
Analysis of postsynaptic densities and morphology
For analysis of the synapse density and morphology, images were taken from randomly sighted stratum lucidum at a magnification of 5,000× for each experimental condition (n=4). The estimation of synapse density was carried out from single photos by counting all synapses (a presynaptic terminal facing a postsynaptic density). For each group, the total pictured area of 1,205~1,507 µm2 was analyzed, and synapse density was expressed as number of synapses per 100 µm2. For determination of the minimal thickness, maximal thickness, length, and total area of each PSD, the NIH software program Image J was used for morphometric measurements.
Western blot analysis
Western blot analysis was performed according to the method reported Suh et al. [36]. Briefly, slices were homogenized to obtain protein extracts. The protein concentration was measured using the Bradford method, and adjusted with 1% SDS. Equal amounts of protein were subjected to 8~10% SDS-PAGE, electrophoretically transferred onto nitrocellulose membranes, and then blocked with 10% skim milk in TBS containing 0.05% Tween 20 (TBST) for 1 h at room temperature. The membranes were then incubated with either mouse anti-PSD-95 (1:1,000; Sternberger Monoclonals, Baltimore, MD), anti-NR1 (1:1,000; Sigma Chemical Corporation, St. Louis, MO), mouse anti-NR2B (1:500; Millipore, Billerica, MA) or mouse-anti-CaMKII (1:1,000; Millipore, Billerica, MA) as a primary antibody for 1 h, and washed three times with TBST (0.05%) for 10 min. Next, the membranes were incubated with a secondary antibody coupled to horseradish peroxidase (HRP) for 45 min. After washing with TBST (0.05%), an enhanced chemiluminescence method (Santa Cruz Biotechnology, Santa Cruz, CA) was used to develop the films. The resulting films were scanned, and densitometric analysis of bands was performed using NIH software program Image J.
Statistical analysis
Data are presented as the mean±standard error of the mean (SEM). Statistical differences between groups were determined by one-way analysis of variance (ANOVA) with Fisher's post-hoc test (Statview 5 version, SAS institute, USA). The threshold for statistical significance was set at p<0.05.
RESULTS
Neuronal cell death was detected in the CA3 area following OGD/reperfusion
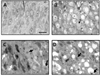
To determine whether neurons are damaged after OGD/reperfusion, toluidine blue staining was performed on the organotypic hippocampal slice cultures. In control slices, the neurons of the CA3 pyramidal cell layer had a normal shape, with large cell bodies and clearly visible Nissl bodies (Fig. 1A). After OGD for 30 min, the cell bodies of the CA3 pyramidal cell layer and adjacent tissue appeared swollen (Fig. 1B). At 6 h (Fig. 1C) and 24 h after reperfusion (Fig. 1D), the number of surviving neurons was decreased, and most remaining neurons showed condensed pyknotic nuclei. Some had a triangular, shrunken perikaryon and were stained dark, suggesting that these neurons were damaged.
OGD/reperfusion induced ultrastructural changes in the stratum lucidum of the CA3 area
In control slices stained with osmium-uranium-lead, CA3 cells had the typical shape of pyramidal neurons, with large round nuclei, homogeneously dispersed karyoplasm, well developed rough endoplasmic reticulum (ER) and polyribosomes, and structurally normal mitochondria (data not shown). Because we targeted the stratum lucidum region of the CA3 area for ultrastructural analysis, there were not many cell bodies present, but many typical dendrites and synapses (presynaptic terminal facing a postsynaptic density). The synapses in control slices were structurally intact with a clear, narrow, synaptic cleft, and many synaptic vesicles located in the presynaptic terminals (Fig. 2A, 2E). After 30 min of OGD, some swelling of the mitochondria, and synaptic terminals was observed. The thickness of the PSD in many synapses became thicker and more electron dense than in controls (Fig. 2B, 2F). At 6 h of reperfusion (Fig. 2C, 2G) and 24 h of reperfusion (Fig. 2D, 2H), the swelling of organelles including mitochondria and neuronal processes, disorganization of the mitochondria and ribosomes, and the thickness and electron density of the PSD were all markedly increased.
The morphology of synapses was remarkably changed in the stratum lucidum of the CA3 area after OGD/reperfusion
In order to evaluate PSD morphology in the CA3 area after OGD/reperfusion, TEM with EPTA staining was performed. After 30 min of OGD, there were no changes on the number, length, thickness and area of PSDs, and those parameters of PSDs were started to be changed at 6 h after OGD (data not shown). At 24 h after OGD, the number of PSDs was significantly decreased by around 50% (13.69±1.21 compared to 27±3.23 in controls, p<0.05, Fig. 3C). The PSD length at 24 h after OGD was significantly increased by approximately 63% (411±13 nm compared to 252±8 nm in controls, p<0.05, Fig. 3D), and the PSD thickness was also significantly increased by approximately 85% (45.1±1.9 nm compared to 24.4±0.7 nm in controls, p<0.05, Fig. 3E).
Levels of PSD proteins decreased after OGD/reperfusion
To investigate the effect of OGD/reperfusion on levels of PSD proteins, western blot analysis for PSD-95, NR1, NR2B, and CaMKII was performed on the hippocampal slice cultures. Levels of all of the analyzed postsynaptic proteins decreased following OGD/reperfusion in a time dependent manner, and showed the lowest levels at 24 h reperfusion after OGD (Fig. 4A & 4B).
DISCUSSION
In the present study, our results show that the morphology of the PSD changed, and that levels of major PSD proteins, such as PSD-95, NR2B, NR1, and CaMKII, were all decreased in the CA3 area of organotypic hippocampal slice cultures following OGD/reperfusion.
Brain ischemia triggers complex cellular mechanisms that impair synaptic functions through the breakdown of cellular and structural features, mediated by various excitotoxic signals [35,37,38]. It is well known that the CA1 area of hippocampus is particularly vulnerable to ischemic insult [39,40]. However, various biochemical and pathological activities, followed by neuronal cell death, can also be detected in the CA3 area along mossy fibers [41,42]. According to our previous studies [31,43], neuronal damage expressed with propidium iodide staining was found both in the CA1 area and CA3 area of hippocampal slice cultures following OGD/reperfusion.
Following OGD for 30 min, some swelling of the mitochondria, ER, dendrites, and synaptic terminals was observed, and the thickness of PSDs was notably increased. During reperfusion, swelling of cellular organelles and disorganization of the mitochondria was evident. This finding is in agreement with that of other studies, which showed some ultrastructural abnormalities in postischemic neurons, including disaggregation of polyribosomes and deposition of dark substances [44-47].
Changes in synaptic responses after variable periods of hypoxia or ischemia have also been demonstrated frequently both in vitro [31,43,48,49] and in vivo [50-52]. In this study, OGD/reperfusion induced modifications as shown by EPTA staining for PSD in the CA3 area. At 24 h after OGD, the number of PSDs was significantly decreased, and the remaining PSDs became thicker, longer, and more electron-lucent, reflecting the damage to PSDs. This finding is consistent with that of earlier studies, which demonstrated that dramatic ultrastructural changes to PSDs, along with synaptic deficits occurred following transient ischemia [35,53-55]. Kovalenko et al. [37] reported a rapid increase in the PSD thickness and length, as well as the formation of concave synapses with perforated PSDs in the CA1 stratum radiatum during the first 24 h after an ischemic episode. EPTA primarily stained the synaptic structure and nucleus, but also stained intracellular protein aggregates. After ischemia, EPTA-stained proteins are predominantly accumulated in rat hippocampal CA1 PSDs, a finding which may be attributable to protein unfolding or denaturing exposing their hydrophobic segments during ischemia, and thereby causing interprotein aggregation in the PSDs after ischemia, and resulting in dysfunctional synaptic transmission at the altered synapse. Additionally, unfolding of proteins induced by ischemia may aggregate and change the structure of PSDs, which may lead to loosening of the PSD frame structure, rendering them more diffuse and irregular [56].
In PSDs, the heteromeric combinations of NR1 and NR2A-D interact with PSD-95 and CaMKII [22,57]. Because this complex plays a central role in the regulation of synaptic function [58-60], and is important in linking the synapse to downstream signaling pathways, it could also be involved in the hypoxia/ischemia-induced changes that lead to neuronal damage. Robust morphological alterations to PSD structure after ischemia/reperfusion were accompanied by biochemical changes, such as decreased levels of heat shock cognate protein 70, CaMKII, and protein kinase C in PSDs [55,61]. When western blot analysis for PSD-95, NR1, NR2B, and CaMKII was performed to investigate the relationship between OGD/reperfusion-induced structural modifications of PSDs, and the PSD protein complex, levels of all the PSD complex proteins were found to be significantly decreased 24 h following OGD (Fig. 5). Previous studies also reported that ischemia/reperfusion [62], or perinatal hypoxia [63] lead to a marked reduction in the mRNA expression of NR1, NR2A, and NR2B, and in the protein expression of PSD-95, total NMDAR levels, and the complexing of PSD-95 with NMDAR subunits within the hippocampal CA1 region, suggesting impaired synaptic performance. Altered PSD-95 expression is centrally involved in the hypoxia-induced changes leading to neuronal injury in the adult brain [55,64].
Taken together, our results show that OGD/reperfusion can induce significant modifications to PSDs in the CA3 area of organotypic hippocampal slice cultures, both morphologically and biochemically. These changes are likely to be part of the process of neuronal cell death and synaptic dysfunction following OGD/reperfusion.




 XML Download
XML Download