

 PDF
PDF ePub
ePub Citation
Citation Print
Print


ABBREVIATIONS
AS
Angelman syndrome
ASD
autism spectrum disorders
CaMKII
Ca2+/calmodulin-dependent protein kinase II
DHPG
dihydroxyphenylglycine
E-LTP
early phase LTP
FMRP
fragile X mental retardation protein
FXS
Fragile X syndrome
HFS
high frequency stimulation
ID
intellectual disability
KO
knock-out
LFS-LTD
low frequecy stimulation LTD
L-LTP
late phase LTP
LTD
long term depression
LTP
long term potentiation
mEPSC
miniature excitatory postsynaptic current
mGluR
metabotropic glutamate receptor
mGluR-LTD
mGluR mediated LTD
NMDA
N-methyl-D-aspartate
PP-LFS
paired-pulse low frequency stimulation
PSD
postsynaptic density
RTT
Rett syndrome
TSC
tuberous sclerosis complex
INTRODUCTION
Synaptic plasticity is frequently measured by analysis of long term potentiation (LTP) and long term depression (LTD) of synapses in different brain regions [1-3]. Historically, the best studied region for synaptic plasticity is the Schaffer collateral pathway in hippocampal CA1 region. The physiological and biochemical mechanisms underlying LTP and LTD have been extensively investigated [3,4]. Over the last decade, analysis of synaptic plasticity has become a popular technique to characterize animal models of neurodevelopmental disorders including autism spectrum disorders (ASD) [5,6]. Various abnormal findings in synaptic plasticity from different brain regions have been reported in mouse models with targeted mutations in genes implicated in ASD (Table 1). However, identifying and interpreting the defects in synaptic plasticity relevant to behavioral manifestations and disease pathophysiology of ASD remain a significant challenge. In this review, we will focus on reviewing the studies of synaptic plasticity in several prominent mouse models for neurodevelopmental disorders with pronounced autistic features and discussing the challenges and future directions in the field.
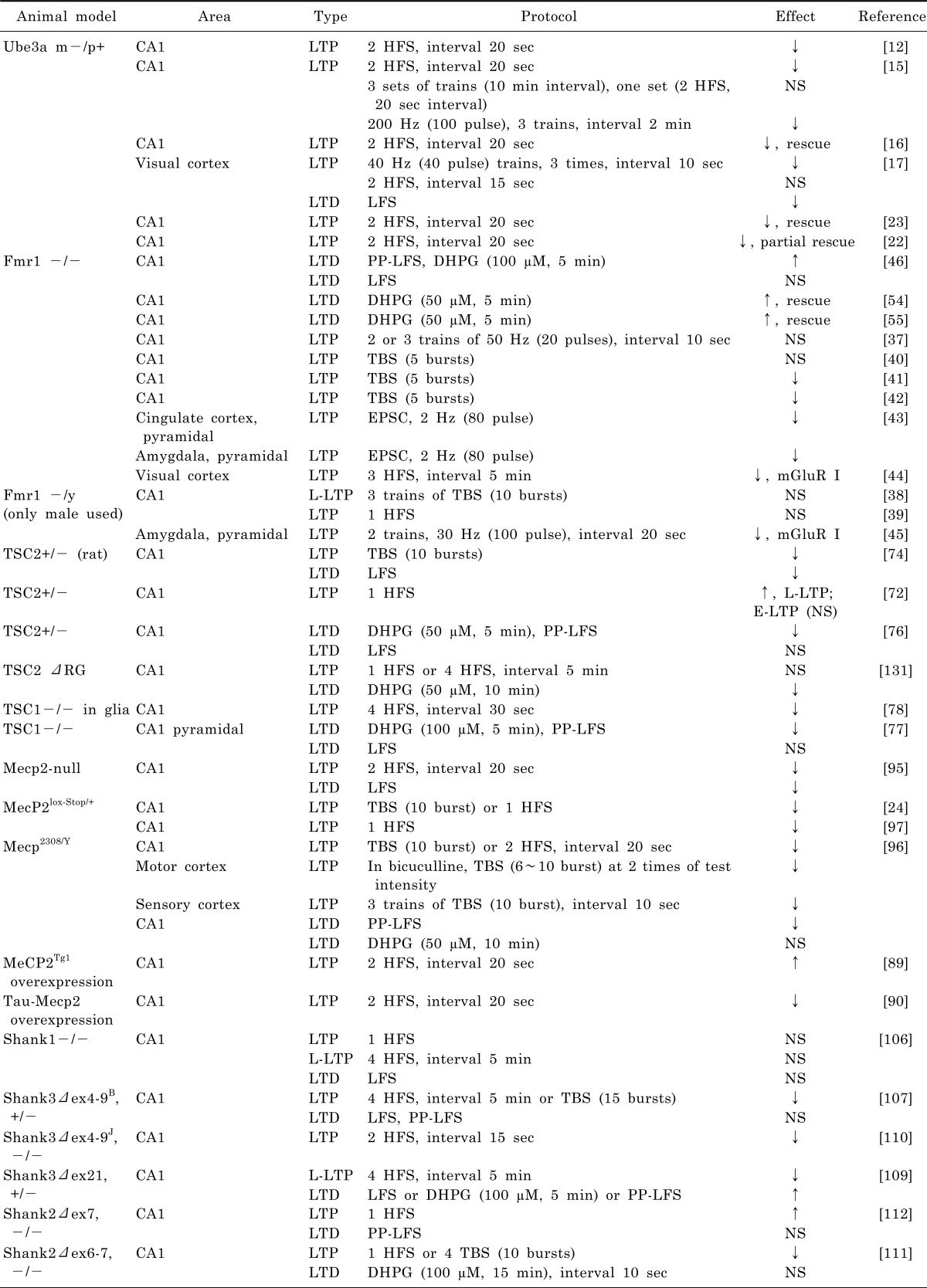
Table 1
Summary of synaptic plasticities with protocols
↓, decrease; ↑, increase; HFS, 100 Hz, 100 pulses; LFS, low frequency stimulation, 1 Hz, 900 pulse; L-LTP, late-LTP; LTD, long-term depression; LTP, long-term potentiation; mGluR I, group I metabotropic glutamate receptor; NS, no significant difference; PP-LFS, paired pulse LFS, paired pulse (50 ms interval), 1 Hz, 900 or 1200 pulse); TBS, theta burst stimulation, 5 to 10 bursts (100 Hz, 4~5 pulse) interval 200 ms. All animals are mice except a note of "rat".
![]()
Go to : 
ANGELMAN SYNDROME
Angelman syndrome (AS) is characterized by profound intellectual disability (ID), movement disorders, absence of speech, epilepsy, and autistic behaviors [7,8]. The molecular defects causing AS include maternal microdeletions on chromosome15q11-q13 (60% of cases), point mutations in the maternal copy of the UBE3A gene (20%), paternal uniparental disomy (5%), and imprinting center defects (1%) [9]. Despite the presence of different molecular defects, it is a well-supported fact that the deficiency of maternal expression of the UBE3A gene in the brain is responsible for the key clinical features of AS [10,11]. To model human AS in mice, the first knock-out (KO) mouse that targeted exon 2 of Ube3a was reported in 1998 and recapitulated the major features of AS in maternal deficiency mice (Ube3a m-/p+) [12]. Subsequently, Ube3a mutant mouse with a mutation in the last coding exon encoding the ubiquitin ligase domain and was reported [13]. In addition, mutant mice with a 1.6 Mb deletion from Ube3a to Gabrb3 that is more similar to AS deletion patients were also reported [14]. However, the Ube3a exon 2 deletion mutant mice have been used more widely by investigators in the research community over the last 15 years.
Synaptic plasticity has been studied extensively in Ube3a m-/p+ mice in different brain regions using different protocols (Table 1). In CA1, LTP is reduced in Ube3a m-/p+ mice using an induction protocol of two trains of high frequency stimulation (HFS) of 100 pulses at 100 Hz (Table 1) [12]. Interestingly, the reduced LTP in CA1 of Ube3a m-/p+ mice could be rescued if a stronger stimulation protocol, three sets of two trains of HFS, was applied [15]. This indicates that the role of Ube3a in synaptic plasticity is probably as a modulator for the expression of LTP and less likely to play an essential role in LTP induction. Unfortunately, LTD at the same synapses has not been investigated so far. In the same study, reduced Ca2+/calmodulin-dependent protein kinase II (CaMKII) activity due to increased inhibitory autophosphorylation was observed in Ube3a m-/p+ mice [15]. In the subsequent rescue experiment, genetic reduction of inhibitory autophosphorylation of CaMKII rescued the LTP deficit as well as hippocampal-dependent learning as assessed by Morris water maze and fear conditioning in Ube3a m-/p+ mice [16]. This in vivo rescue indicates that a biochemical mechanism mediated by CaMKII activity underlies the impaired synaptic plasticity in Ube3a-deficient synapses. However, it remains unclear what is the molecular mechanism through which the deficiency of Ube3a contributes to the altered activity of CaMKII.
Synaptic plasticity was also investigated in visual cortex in Ube3a m-/p+ mice [17,18]. Both LTP and LTD are reduced in visual cortex in Ube3a m-/p+ mice (Table 1) [17]. Low frequency stimulation was used for N-methyl-D-aspartate (NMDA) receptor-dependent LTD (LFS-LTD) induction but group I metabotropic glutamate receptor (mGluR) mediated LTD (mGluR-LTD) was not analyzed. Interestingly, in vitro synaptic plasticity was affected by the in vivo visual experience of the mice. Deprivation of visual input achieved by a dark rearing environment could actually restore the impaired LTP and LTD in Ube3a m-/p+ mice [17]. This observation is intriguing because it indicates that the defective synaptic plasticity due to deficiency of Ube3a can be easily reversed in animals. On the other hand, it would be interesting to learn whether dark rearing may actually reverse abnormal behavioral phenotypes in Ube3a m-/p+ mice.
As the UBE3A gene is a brain-specific imprinted gene that is expressed primarily from the maternal chromosome, the UBE3A gene on the paternal chromosome is structurally intact but transcriptionally repressed [9,19]. Through a small molecule screening program, Huang et al. discovered that the silenced Ube3a from the paternal chromosome can be activated by topotecan, a toposiomerase inhibitor, in vitro and in vivo in mice [20]. This discovery opens an exciting research avenue to explore the treatment of AS using pharmacological interventions and presumably through epigenetic modification [21]. The immediate question is whether postnatal treatment with toposomeriase inhibitors can rescue the synaptic plasticity and ultimately behavioral defects in Ube3a m-/p+ mice. Another approach of treatment is to use a virus-mediated gene delivery method in hippocampus in vivo. Restoring the expression of Ube3a could rescue the early phase of LTP impairment and cognitive deficits in Ube3a m-/p+ mice [22]. In yet another group, use of an ErbB inhibitor could rescue LTP in Ube3a m-/p+ [23]. These observations together suggest that impaired synaptic plasticity and behavioral abnormalities in Ube3a m-/p+ mice is reversible even during late development. A similar finding was also reported in Mecp2 mutant mice [24,25]. However, it remains a subject of debate if the same phenomenon may exist in the treatment of human neurodevelopmental disorders.
A subset of AS patients meet the diagnostic criteria for ASD [26]. However, the behavioral studies of Ube3a m-/p+ mice have not revealed significant impairments in ASD-like behaviors although these mice have recapitulated the most salient features of intellectual disability and movement disorder seen in AS [27]. This result may indicate that other genes in the 15q11-q13 region may also contribute to the ASD in human AS. Alternatively, more extensive behavioral tests and comparisons between mutant mice with mutation only in Ube3a and a deletion from Ube3a to Gabrb3 may be warranted [14].
Go to :
FRAGILE X SYNDROME
Fragile X syndrome (FXS) is one of the best studied disorders with intellectual disability ID and ASD both in humans and mouse models [28]. In addition to ID and ASD, FXS patients are characterized by seizures, macroorchidism, and dysmorphic facial features [29]. Molecularly, FXS is caused by the CGG triplet expansion in the 5' untranslated region of the FMR1 gene on the X chromosome [30,31]. The expanded CGG repeat results in promoter methylation that represses the transcription of FMR1 [32]. The FMR1 gene encodes the fragile X mental retardation protein (FMRP), an RNA-binding protein which inhibits local protein translation stimulated by group I mGluR signaling [28,33,34]. To understand the pathogenesis of fragile X syndrome, Fmr1 KO mutant mice were first developed in 1994 [35]. For almost two decades, Fmr1 mutant mice have been extensively studied from many angles by numerous investigators [36]. The full review of the findings from studying Fmr1 mutant mice is beyond the scope of this review. Instead, we will focus on synaptic plasticity in Fmr1 mutant mice.
The initial studies of synaptic plasticity in hippocampal CA1 region in Fmr1 mutant mice did not reveal any impairment in LTP by a standard LTP induction protocol (Table 1) [37-40]. However when the stimulation for LTP induction was reduced to near the induction threshold level in subsequent studies, LTP in CA1 was found to be reduced in Fmr1 KO [41,42]. Interestingly, LTP in brain regions including somatosensory cortex, anterior cingular cortex, anterior piriform cortex and lateral amygdala was also found to be decreased [39,40,43]. Impaired LTP was also observed in visual cortex and basolateral amygdala, but notably they were mGluR dependent [44,45]. These observations indicate different region- or synapse-specific defects in Fmrp deficient mice. However, the most important finding from synaptic plasticity studies is the observation of enhanced mGluR-LTD in hippocampal CA1 region [46]. A similar phenomenon of enhanced LTD was observed in cerebellum [47]. The observation of enhanced LTD in hippocampal CA1 region led to a theory of aberrant mGluR signaling underlying the pathophysiology of FXS [48]. The central hypothesis of the mGluR theory is that loss of FMRP in the synapse leads to the up-regulation of the mGluR-mediated signaling pathway. The mGluR theory and the molecular mechanism underlying the enhanced mGluR-LTD have been tested extensively since it was proposed and these studies have validated the central hypothesis [46,49-51]. However, alterations of many signaling pathways and a long list of potential protein targets in synapses have been revealed in Fmr1 mutant mice [28,52]. It is not entirely clear how the disruption of these different pathways can be integrated into a unifying mechanism responsible for the pathophysiology of FXS. Recent reports indicate an involvement of Homer proteins in the dysregulated mGluR signaling pathway [53]. Genetic reduction of mGluR5 or pharmacological inhibition of mGluR5 could rescue the abnormal behaviors in Fmr1 mutant mice [54,55]. Similarly, genetic reduction of Homer1a in Fmr1 KO could also improve behaviors, though this did not rescue mGluR-LTD in hippocampus [56]. These rescue experiments raise an interesting possibility for potential reversal of neurological impairments in human fragile X syndrome. The various synaptic defects found in different brain regions in Fmr1 mutant mice raise an immediate question about the correlation between the defective synaptic plasticity and the abnormal behaviors for future investigation. For example, social behaviors are impaired in Fmr1 KO mice which is consistent with autistic behaviors frequently seen in human fragile X syndrome patients [57-60]. These studies support Fmr1 KO mice as a good model to dissect the pathophysiology and explore treatment strategies for ASD [28].
Go to :
TUBEROUS SCLEROSIS COMPLEX
Tuberous sclerosis complex (TSC) is a neurocutaneous condition with prominent neurobehavioral manifestations including seizures, ID, and autistic behaviors [61,62]. The neurobehavioral features are quite variable and range from mild to severe presentations in TSC patients [61]. TSC is caused by mutations in TSC1 or TSC2 genes that show a dominant inheritance pattern [63,64]. The proteins, hamartin encoded by TSC1 and tuberin encoded by TSC2 genes, form a heterodimeric complex that functions as a negative regulator for the mTOR pathway [65-67]. Therefore, it has been hypothesized that loss of function mutations in TSC1 or TSC2 disinhibit mTOR signaling and lead to the up-regulation of the signaling pathway downstream of mTOR which promotes cell growth and proliferation [67,68].
Both homozygous Tsc1 or Tsc2 KO mice are embryonic lethal [69-71]. Heterozygotes of Tsc1 or Tsc2 mutation exhibit cognitive impairment and synaptic dysfunction in the absence of apparent neuroanatomical defects or seizures [72-75]. In Tsc2+/- rats (Eker rat), LTP and LFS-LTD was decreased in CA1 [74]. In Tsc2+/- mice, early phase LTP (E-LTP) in hippocampal CA1 is not affected but late phase LTP (L-LTP) was enhanced [72]. In Tsc2+/-, mGluR-LTD was decreased but LFS-LTD was intact [76]. The reduced mGluR-LTD in Tsc2+/- is opposite to what is seen in Fmr1 KO mice although the mGluR-LTDs from both were insensitive to protein synthesis inhibitors [76]. As in Tsc2+/-, Tsc1+/- mutant mice showed a similar impairment in synaptic plasticity. In hippocampal CA1 pyramidal neurons with conditionally deleted Tsc1, mGluR-LTD was reduced but LFS-LTD was intact [77]. Interestingly, synaptic plasticity is also impaired when Tsc1 was knocked out in non-neuronal cells. For instance, the E-LTP was reduced in Tsc1 glia-specific conditional KOs [78]. A recent study on Tsc1 deleted specifically in cerebellar Purkinje cells showed impaired social interaction, enhanced repetitive behaviors and abnormal ultrasonic vocalizations [79]. However, synaptic plasticity was not tested in cerebellum in this mouse model [79]. This observation raises a provocative question regarding the brain regions and circuits that are important for the pathophysiology of autistic behaviors because social interaction was significantly reduced both in Tsc1+/- and Tsc2+/- [73,79,80]. The advantage of TSC models over other ASD mouse models is that the signaling pathway involving dysregulation of mTOR is well defined in both Tsc1 and Tsc2 mutant mice.
Go to :
RETT SYNDROME
Rett syndrome (RTT) is a neurological disorder that primarily affects females and is caused by mutations in the MeCP2 gene [81,82]. The clinical presentations of RTT are characterized by normal early neurodevelopment for the first 12~18 months followed by developmental regression [83]. The major symptoms of RTT include movement disorders, absence of speech, and repetitive hand movements [83]. MeCP2 protein generally is considered to suppress transcription by binding to methylated CpG DNA [84]. However, recent evidence suggests a role of bidirectional regulation with both repression and activation of transcription mediated by MeCP2 [85]. Several Mecp2 mutant mice carrying slightly different mutations have been produced and characterized [24,86-88]. In addition, mutant mice with overexpression of Mecp2 was also reported [89,90]. These mice are valuable models to ASD research because RTT is a prototype for syndromic ASD and because impairments in social behaviors were observed in both whole brain- and region specific Mecp2 mutant mice [91-94].
In general, mice lacking the functional copy of Mecp2 recapitulate the major features of RTT. In Mecp2-null mouse, synaptic plasticity was analyzed at two different ages because of the age-dependent regression in human RTT [86,95]. In male mice at a presymptomatic age (3~5 weeks old), no difference in LTP at hippocampal CA1 region was found. However, at a symptomatic age (6~10 weeks old) LTP and LFS-LTD in CA1 was reduced. This indicates that the trajectory of impaired synaptic plasticity correlates well with the developmental phenotype changes as suggested in humans. In a model where Mecp2 was truncated as in some human patients (Mecp2308/Y), LTP and paired-pulse low frequency stimulation (PP-LFS) (Table 1) but not the group I mGluR agonist dihydroxyphenylglycine (DHPG)-LTD was reduced in hippocampal CA1 from male mice [87,96]. Similarly, LTP in primary motor cortex and sensory cortex was reduced [96]. More interestingly, the impaired LTP in CA1 and neurological phenotypes in Mecp2lox-Stop/+ mice could be rescued by reintroduction of Mecp2 by genetic manipulation in male or female mice [24,97]. At least in three different RTT models, decreased CA1 LTP was consistent.
Synaptic plasticity was also investigated in mice with overexpressed MeCP2 via BAC mediated transgenics (MeCP2Tg1) [89]. As predicted from the finding of reduced LTP in Mecp2 deficiency mice, LTP in CA1 was increased in MeCP2Tg1 [89]. However LTP in CA1 was decreased in a different animal model with the overexpression of Mecp2 driven by Tau promoter in neurons (Tau-Mecp2) [90]. The explanation for this discrepancy is not apparent and additional investigation is warranted.
Go to :
SHANK FAMILY GENE CAUSING ASD
SHANK family proteins, SHANK1, SHANK2, and SHANK3, are scaffolding proteins enriched at the postsynaptic density (PSD) of excitatory synapses [98]. SHANK proteins share a similar protein domain structure that mediates protein-protein interaction at the PSD for synaptic function [98,99]. Molecular defects in SHANK3 were first found in patients with ASD and ID [100]. Subsequently, genetic defects of SHANK1 and SHANK2 were also reported in ASD and ID [101-103]. Because of the existing knowledge of the function of SHANK family proteins at synapses, the discovery of mutations in SHANK family genes provide direct support for the notion that the pathogenesis of ASD may reside in the dysfunction of synapses [104,105]. Mutant mice for all Shank family genes have been reported [106-112]. Shank1 mutant mice were first reported [106]. Surprisingly, the phenotype of Shank1 deficiency mice was unexpectedly mild. No synaptic plasticity changes were detected in LTP and LFS-LTD in the Shank1 KO even though mEPSC frequency and synaptic strength were decreased [106]. Because of the findings of both microdeletions of and point mutations in the SHANK3 gene in human ASD [100,113], the interest to model Shank3 mutations in mice has been intensified recently. This led to the simultaneous generation of multiple Shank3 mutant mice by disrupting different portions of Shank3 exons [107-110]. These mutations include deletion of exons 4-7 (Δex4-7) [108], exons 4-9 (Δex4-9J) [110] and (Δex4-9B) [107], exon 11 (Δex11) [112], exons 13-16 (Δex13-16) [108] and exon 21 (Δex21) [109]. We recently discovered that Shank3 has an array of protein isoforms resulting from the combination of multiple intragenic promoters and extensive alternative splicing of coding exons [110]. Therefore, we concluded that different mutations in different exons resulted in the disruption of different Shank3 isoforms but none of these mutant mice were Shank3 complete knockouts. Shank proteins regulate the abundance and signaling of ionotropic and metabotropic glutamate receptors at excitatory synapses [105,114]. Accordingly, synaptic transmission and plasticity were examined in different brain regions in all Shank3 mutant mice. Measurements of miniature excitatory postsynaptic current (mEPSC) frequency and amplitude, paired pulse ratio, input/output (I/O) curves, fiber volley, and population spikes indicated that synaptic transmission was reduced at hippocampal CA1 synapses of Δex4-9B+/- mice [107], but not in mice bearing Δex4-9J-/- [110], Δex13-16-/- [108], or Δex21+/- mutations [109]. The explanation for the difference between Δex4-9B+/- and Δex4-9J-/- is not immediately clear.
In striatum, the frequency of mEPSCs and amplitude of population spikes were significantly decreased in Δex13-16-/- mice, but only mildly affected in Δex4-7-/- mice [108]. Presynaptic responses measured by paired pulse ratio and input/output curves were not altered at corticostriatal synapses in Δex13-16-/- or Δex4-7-/- mice [108]. The different degree of synaptic transmission defects in mice with specific Shank3 mutations supports the notion of an isoform-specific contribution to synaptic function. Moreover, the reduced NMDA receptor-mediated responses at cortical synapses of Δex21+/- [109] but not in the corticostriatal synapses of Δex13-16-/- mice [108] indicate distinct functions of Shank3 at different synapses.
In terms of plasticity, hippocampal LTP was reduced at CA1 synapses of Δex4-9J-/-, Δex4-9B+/-, and ex21+/- mice [107,109,110]. In contrast, LFS-LTD was reduced in CA1 of Δex21+/- mice [109] but not in Δex4-9B+/- mice [107], suggesting an alteration in the set-point for bidirectional Hebbian synaptic plasticity [115]. mGluR-LTD induced by DHPG or PP-LFS was enhanced in CA1 hippocampus of Δex21+/- mice. However, a similar enhancement of mGluR-LTD was not evident in the Δex4-9B+/- mice induced by PP-LFS [107]. In addition, mGluR1/5 protein levels were not altered in Δex21+/- mice [109].
Collectively, these data support synaptic defects mediated by glutamate receptors in Shank3 mutant mice that appear to be both synapse- and mutation-specific. It is not yet clear whether there are common core circuit defects in the various mutant mice, but the phenotypic heterogeneity itself appears consistent with the clinical heterogeneity of patients harboring SHANK3 mutations. Since different mutations affect different isoforms of Shank3, some of the observed phenotypes may arise from isoform-specific effects on synaptic transmission.
Two mutant models for Shank2 were reported recently [111,112]. Schmeisser et al. reported Shank2 exon 7 deletion mutant mice (Shank2 Δex7) in which LTP in hippocampal CA1 was increased but no change in LTD with PP-LFS was observed [112]. Reduced social interaction, increased stereotypical behavior, hyperactivity, and altered ultrasonic vocalization pattern were found in Shank2 Δex7 -/- mice. Won et al. generated Shank2 mutant mice where exons 6-7 were deleted (Shank2 Δex6-7) [111]. Both exon 7 and exon 6-7 deletion resulted in a frame shift mutation shortly after exon 7. Intriguingly, LTP in hipppocampal CA1 was reduced in Shank2 Δex6-7 mice and this is opposite to Shank2 Δex7 mice [111]. In addition, the NMDA current and LFS-induced LTD were reduced in hippocampal CA1 region in Shank2 Δex7 mice but DHPG-induced LTD was not affected. The behavioral profile of Shank2 Δex6-7 mice is very similar to Shank2 Δex7. Interestingly, treatment with NMDA agonist current mediated signaling could rescue social interaction deficits [111]. The explanation for the apparent discrepancy in synaptic plasticity but similar behavioral profile in mice with two very similar mutations is not immediately clear and further investigation is warranted. However, available data strongly support that both Shank2 and Shank3 mutant mice are valid ASD models to dissect the pathophysiology.
Go to :
FUTURE DIRECTIONS AND CHALLENGES
It is clear that abnormalities in synaptic plasticity vary significantly among different animal models of ASD. For instance, Tsc2 mutations in rat and mice have opposite effects on LTP [72,74]. Shank1 mutant mice do not show plasticity defects using standard protocols unlike Shank2 and Shank3 mutants (Table 1). In Shank2 mutants, LTP impairment is in opposite directions in two different lines of mutant mice despite similar mutations and behavioral profiles [111,112]. However, as an example of convergence, LTP in three different lines of Shank3 mutants from three different groups was decreased consistently but the LTD defects are significantly different [107,109,110]. On the other hand, it is difficult to correlate the abnormal plasticity with the corresponding behavioral manifestations in each model.
Currently, most synaptic plasticity experiments were performed in the hippocampus while the deficiency of targeted genes was in the whole brain. Therefore, it is difficult to establish causality between brain region and abnormal behaviors studied in the models. Because the neuroanatomical basis for autism is still poorly understood, an unbiased survey for synaptic plasticity in other different brain areas may provide more informative data about the pathophysiology of autism.
In human brain, the superior temporal sulcus region, the fusiform gyrus and amygdala are considered important for social interaction and gaze behaviors [116]. However, gaze behavior in mice, which are nocturnal, is difficult to monitor technically. A neural circuit involving amygdala could be important region to study in ASD mouse model. The study of amygdala and related circuits such as medial prefrontal cortex in autism mouse models is within reach [117,118].
For stereotypical behaviors, cortical-striatal circuits are hypothesized to be important in ASD [119-121]. Significant repetitive behaviors measured by increase in self-grooming and inflexibility in the reversal phase of the Morris water maze are frequently reported in ASD mouse models [107,108]. The synaptic plasticity in cortical-striatal circuit activity is less well characterized in these most ASD models [122].
For the aspect of communication/language impairment in humans, ultrasonic vocalization (USV) recording in mice has become a popular approach despite ongoing debate about the value of the USV relevant to human communication [123,124]. Abnormal USV measurements have been reported in numerous ASD mouse models [14,108-112,125-128]. These observations support the value of USV recording because of easy quantification and detailed numerical analysis. However, several challenges remain. First, what is the ethological meaning of USV in mice? Second, what is the circuit in rodent brain responsible for USVs [129,130]. More investigation are clearly warranted in future.
The studies of synaptic plasticity and behaviors in these high profile mouse models with defined genetic defects have produced many interesting findings but also raise numerous challenging questions. First, what is the implication of variable, or opposite in some cases, synaptic plasticity related to understanding the pathophysiology of ASD and other comorbidities? Second, what is the molecular mechanism underlying the different synaptic plasticity between different brain regions? Third, can impaired synaptic plasticity in a particular brain region predict abnormal behaviors? Fourth, which is a more reliable biomarker, the synaptic plasticity or behavioral defects, to use for future drug screening? Future investigations may focus on 1) generation of brain region-, cell type-, or circuit-specific targeted gene KO, 2) in vivo physiology or circuit analysis, and 3) development of new and sensitive behavioral tests. Despite these challenges, we have reasonable confidence that studying these and other new ASD models will lead to better understanding the pathophysiology of ASD and ultimately lead to the development of new treatments.
Go to :
 XML Download
XML Download