

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Transient receptor potential vanilloid subtype 1 (TRPV1) is a ligand-gated, non-selective cation channel that is activated by noxious heat (>43℃), acids, and capsaicin [1-3]. Based on reports that TRPV1 knock-out mice exhibit reduced inflammatory thermal hyperalgesia, TRPV1 appears to be essential for mediating thermal hyperalgesia induced by inflammation [4,5]. TRPV1 activity is modulated by various substances involved in inflammation, such as bradykinin [6], histamine [7], prostaglandins [8], and the proinflammatory chemokine CCL3 [9]. By activating or inhibiting kinases (e.g., protein kinase A, protein kinase C, and phosphatidylinositol 3 kinase), these inflammatory mediators modulate TRPV1 responses to stimuli [10]. Additionally, TRPV1 activity can be modulated by changes in the redox state of the cellular environment in dorsal root ganglion (DRG) neurons and human embryonic kidney (HEK) cells [11-13]. These reports suggest that TRPV1 is susceptible to being modulated by various factors related to inflammation.
Nitric oxide (NO) has been recognized as an important signaling molecule in various physiological processes, including apoptosis, inflammation, and synaptic transmission [14-16]. NO is a gaseous mediator that is synthesized from L-arginine in a reaction catalyzed by a family of nitric oxide synthase (NOS). NO signaling occurs via at least two separate pathways. In the first, NO stimulates soluble guanylyl cyclase (sGC) to increase cyclic guanosine monophosphate (cGMP), which, in turn, modulates various downstream signaling targets. In the second pathway, NO covalently and reversibly forms nitrosothiols (R-S-N=O) with free thiols of cysteine residues within proteins and hence directly modifies protein function [17]. It has been reported that the activities of various voltage- and ligand-gated ion channels are modulated by NO, through direct interactions with protein structures [18-21] and indirect mechanisms, involving cGMP-dependent protein kinase (PKG) activation [22-27].
Recently, it was reported that NO donors could directly activate TRPV1 by S-nitrosylation, which involves the transfer of a NO group to cysteine sulfhydryl residues on the protein [28,29]. There are several reports that NO can modulate cellular function via both direct and indirect sGC/PKG pathways [30-33]. Thus, we were interested in whether NO could modulate the activity of TRPV1 through the sGC/PKG pathway. In this study, we investigated the modulation of TRPV1 activity via the indirect cGMP/PKG pathway using whole-cell patch clamp recordings.
Go to : 
METHODS
Cell culture
Primary cultures of DRG neurons dissected from all levels of the lower cervical, thoracic, and lumbar spinal cord of 2-day-old rats were prepared. The vertebral column was cut along the centerline and spread to both sides to expose DRGs near the bottom. Under a dissecting microscope, DRGs were removed from both sides of the spinal column with forceps. The dissected ganglia were collected in cold (4℃) culture medium, consisting of a Dulbecco's modified Eagle's medium/F-12 mixture (DMEM/F12; Gibco, Invitrogen, Grand Island, NY, USA), 10% fetal bovine serum (FBS; Gibco, Invitrogen), 1 mM sodium pyruvate, and 100 U/ml penicillin/streptomycin (Sigma-Aldrich, St. Louis, MO, USA). The collected ganglia were washed with culture medium and incubated at 37℃ for 30 min in 1 mg/ml collagenase (Type II; Worthington, Freehold, NJ, USA). The ganglia were then washed three times with Mg2+- and Ca2+-free Hank's balanced salt solution (HBSS; Gibco, Invitrogen) and incubated in 2.5 mg/ml trypsin (Gibco, Invitrogen) at 37℃ for 30 min. Subsequently, the ganglia were centrifuged (1,000 rpm, 10 min) and the pellet was washed two or three times with culture medium to inhibit the enzyme. The pellet was then resuspended in culture medium by gentle trituration with a Pasteur pipette, and the suspended cells were plated on square glass coverslips coated with poly-L-lysine (Sigma-Aldrich), which were then placed in small Petri dishes. Then, nerve growth factor (25 ng/ml; Alomone Labs, Jerusalem, Israel) was added to each Petri dish. Cells were incubated at 37℃ in a 95% air-5% CO2 gas mixture and were used 1~3 days after plating.
Electrophysiology
Whole-cell currents were recorded from the somata of small-diameter DRG neurons. Electrodes were made by pulling borosilicate glass capillaries (Harvard, Kent, UK). Tip resistances were 2~3 MΩ for whole-cell recordings. To record whole-cell currents, the cell membrane was ruptured by gentle suction after the formation of a gigaohm seal. Capacitative transients were then cancelled. Whole-cell membrane currents were recorded at -60 mV using an Axopatch 200B amplifier (Molecular Devices, Union City, CA, USA). Membrane currents were low-pass filtered at 1 kHz and sampled at 2.5 kHz with a Digidata 1322 data acquisition system (Molecular Devices). Data were analyzed using the pClamp 8.0 software (Molecular Devices). All electrophysiological experiments were performed at room temperature.
Solutions and chemicals
The Ca2+-free bath solution contained 140 mM NaCl, 5 mM KCl, 10 mM HEPES, 10 mM glucose, 1 mM MgCl2, and 10 mM EGTA (pH adjusted to 7.4 with NaOH). Recording pipettes were filled with an internal solution containing 110 mM K gluconate, 1 mM MgCl2, 10 mM NaCl, 10 mM BAPTA-K4, 0.5 mM EGTA, 10 mM HEPES, 2 mM Mg-ATP, and 0.1 mM Na-GTP, pH 7.4 (adjusted using KOH).
All chemicals except 2-(4-carboxyphenyl)-4,4,5,5-tetramethylimidazoline-1-oxyl-3-oxide (CPTIO; Tocris Bioscience, Bristol, UK) were purchased from Sigma-Aldrich. Capsaicin was dissolved and stored as a 10 mM stock solution in 100% ethanol. S-nitro-N-acetylpenicillamine (SNAP, 50 mM), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10 mM), and bisindolylmaleimide (BIM, 10 mM) were dissolved in dimethyl sulfoxide (DMSO) as stock solutions, and 3-isobutyl-1-methylxanthine (IBMX, 1 mM) and KT5720 (1 mM) were prepared as methanol stock solutions. KT5823 was dissolved as a 10 mM stock solution in ethyl acetate. All stock solutions were stored at -20℃ and were thawed immediately before use; they were then diluted with the bath solution to the desired final concentrations at the beginning of each experiment.
Statistical analyses
Data are presented as means±standard error of the mean (SEM). All comparisons between means were tested for significance using a Student's paired t-test. p values<0.05 were considered to indicate statistical significance.
Go to :
RESULTS
NO donors decreases capsaicin-activated currents
We first tested the effects of the extracellular application of NO donors on capsaicin-activated inward currents (Icap) in cultured rat DRG neurons (Fig. 1). When capsaicin (0.3 µM) was applied in the bath for 20 s, an inward membrane current was developed within a few seconds, due to opening of TRPV1. Because Icap can be diminished by repetitive applications of capsaicin in a Ca2+-dependent manner [34], we used Ca2+-free bath solution and a pipette solution containing 10 mM BAPTA to avoid such tachyphylaxis throughout the experiments. After membrane currents were recovered to resting levels by washing out capsaicin with bath solution, sodium nitroprusside (SNP, 100 µM) was co-applied with capsaicin. The amplitude of Icap+SNP decreased significantly, to 41.9±1.6% of the control Icap (p<0.01, n=22). This decrease was reversible; the magnitude of Icap recovered spontaneously to 74.5±3.9% of the control capsaicin response after the SNP had been washed out (Fig. 1A). The decreasing effect of SNP co-applied with capsaicin (45.5±1.6% of the control) was not significantly different from that of SNP pretreatment for 1 min before capsaicin application (40.7±8.0% of the control, n=3, data not shown). The NO donor SNAP (100 µM) also significantly decreased Icap to 30.8±1.4% of the control response (p<0.01, n=13; Fig. 1B). In the dose-response analysis, the IC50 of SNP for TRPV1 inhibition was 0.67 µM (Fig. 1C). These results suggest that NO donors decrease the amplitude of Icap in a reversible and dose-dependent manner.
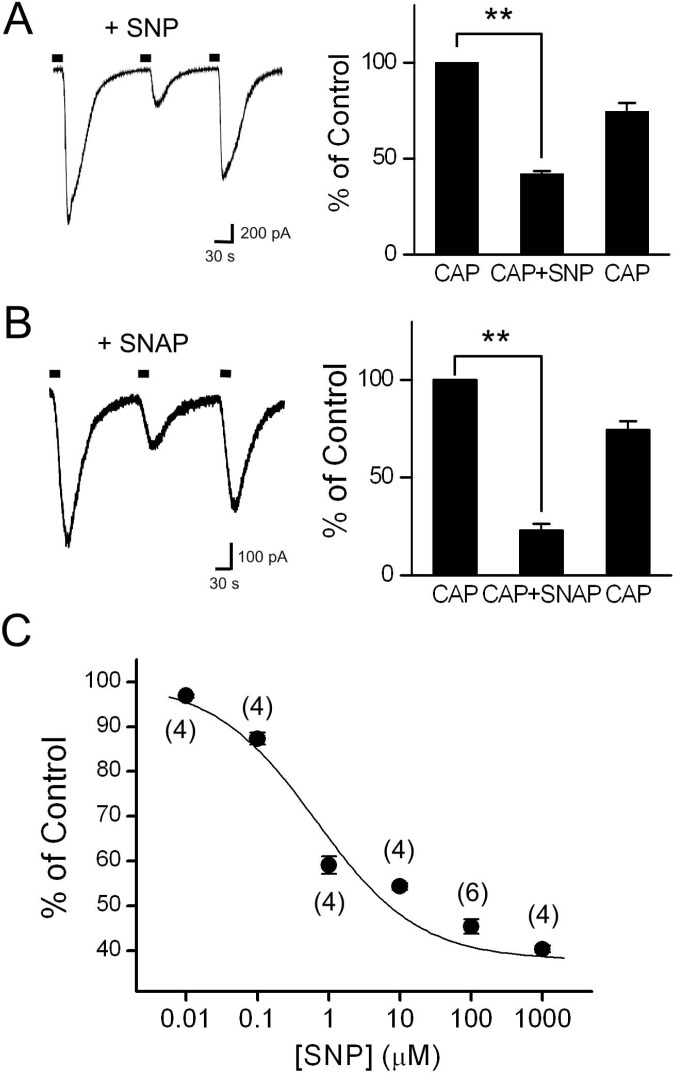 | Fig. 1Effects of the NO donors, SNP and SNAP, on Icap in cultured DRG neurons. (A) Application of SNP (100 µM) with capsaicin (CAP, 0.3 µM) decreased Icap significantly (n=22). The representative trace shows Icap in the control and during application of SNP at a holding potential of -60 mV. Filled bars on the trace indicate the application of capsaicin for 20 s. The bar graph summarizes the effects of SNP on Icap. (B) SNAP (100 µM) co-applied with capsaicin also inhibited Icap significantly (n=13). The bar graph summarizes the effect of SNAP on Icap. (C) Dose-response curve for SNP on Icap inhibition. The curve was fitted using the Hill equation (IC50=0.67 µM). Data are expressed as a percentage of the control capsaicin response, represented by Icap in the absence of SNP, and are plotted as means±SEM. Numbers in parentheses indicate the number of cells tested. Asterisks indicate significant differences relative to the control, at **p<0.01.
|
NO scavengers prevent diminution of Icap by NO
We next investigated the effects of NO scavengers to determine whether the decrease in Icap by SNP was due to the release of NO from the donor. SNP (100 µM) decreased the amplitude of Icap by 56.8±1.7% (p<0.01; Fig. 2A). In the same cells, SNP reduced the amplitude of Icap only by 5.5±1.4% (p<0.01, n=7) in the presence of hemoglobin (Hb, 20 µM). When cells were pretreated with a specific NO scavenger, CPTIO (200 µM), subsequent application of SNP also failed to decrease Icap (percentage inhibition was 3.9±1.5%, p<0.01, n=7; Fig. 2B, C). Elimination of the SNP-mediated inhibition of Icap by NO scavengers confirmed that NO released from the donors played a role in Icap inhibition.
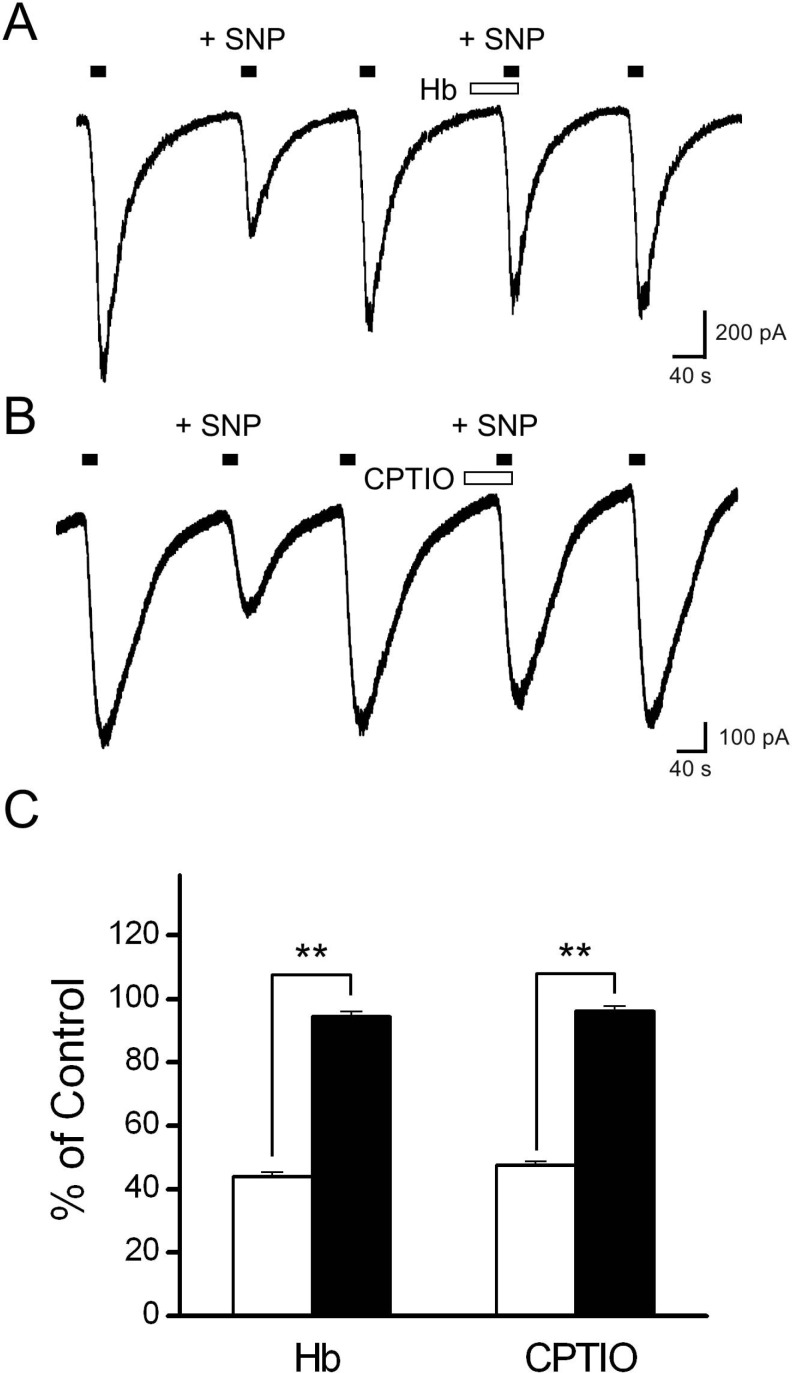 | Fig. 2Effects of the NO scavengers, hemoglobin and carboxy-PTIO, on SNP inhibition of Icap. (A) The representative trace shows Icap during the control, application of SNP, and application of SNP in the presence of hemoglobin (Hb). Hemoglobin eliminated the inhibition of Icap caused by subsequent application of SNP. (B) The representative tracing shows Icap during the control, application of SNP, and application of SNP in the presence of carboxy-PTIO. Carboxy-PTIO also prevented the inhibition of Icap caused by subsequent application of SNP. Filled bars on the trace indicate the application of capsaicin for 20 s, and the open bar represents the application of the NO scavengers. (C) The bar graph summarizes the effects of SNP on Icap in the presence (filled bar) and absence (open bar) of the NO scavenger. Data are presented as means±SEM. Asterisks indicate significant differences at **p<0.01.
|
Involvement of guanylyl cyclase and cGMP in the Icap reduction caused by SNP
NO has been shown to activate sGC, leading to an increase in cGMP levels (for review, see [35]). Thus, we hypothesized that if the effect of NO was mediated by the activation of sGC, then inhibitors of sGC would block the inhibitory effect. First, ODQ, a selective sGC inhibitor, was tested to determine whether SNP inhibition of Icap was associated with activation of sGC. When ODQ (10 µM) was pretreated prior to capsaicin and SNP application, the inhibitory effect of SNP was alleviated (percentage inhibition: 19.8±2.1%, p<0.01, n=6; Fig. 3A, E).
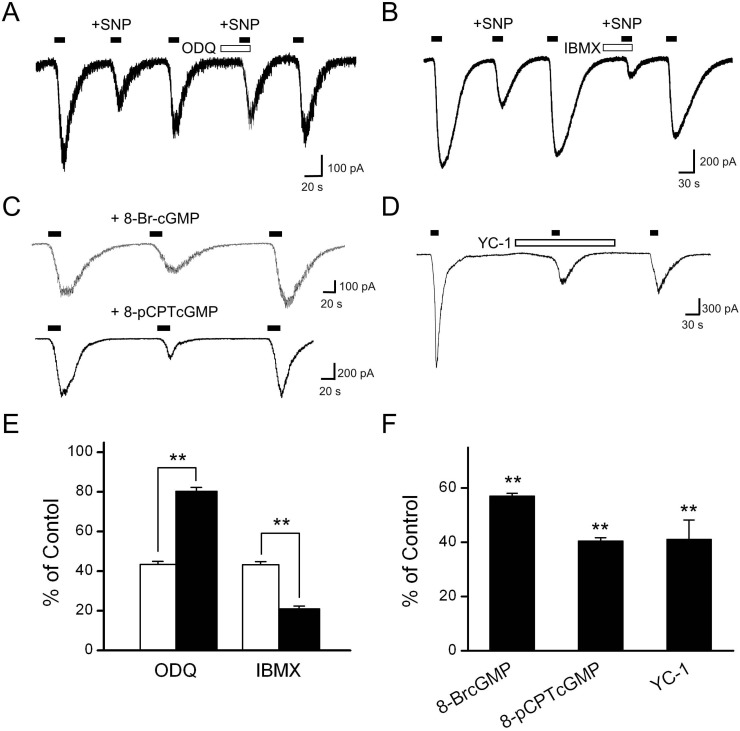 | Fig. 3Involvement of guanylyl cyclase and cGMP in the effects of SNP on Icap. (A) The representative trace shows Icap during the control situation, the application of 100 µM SNP, and the application of SNP in the presence of 10 µM ODQ. Pretreatment with ODQ partially blocked the inhibitory effect of SNP on Icap (n=6). (B) The representative trace shows Icap during the control situation, the application of 100 µM SNP, and the application of SNP in the presence of 1 mM IBMX, a phosphodiesterase inhibitor. The inhibitory effect of SNP was augmented by pretreatment with IBMX (n=9). Filled bars on the trace indicate the application of capsaicin for 20 s, and the open bar indicates the application of the inhibitor. (C) Each representative trace shows the effects of membrane-permeable analogs of cGMP, 8-Br-cGMP (100 µM) and 8pCPT-cGMP (100 µM), on the capsaicin responses. Both 8-Br-cGMP and 8-pCPT-cGMP mimicked the inhibitory effect of SNP on Icap. (D) The NO-independent guanylyl cyclase activator YC-1 (30 µM) decreased Icap. (E) Summary data show the effect of SNP on the amplitude of Icap before and after treatment with ODQ and IBMX. (F) The bar graph summarizes the effects of the membrane-permeable analogs of cGMP and YC-1 on Icap. Data are presented as means±SEM. Asterisks indicate significant differences at **p<0.01, vs. control.
|
cGMP is broken down to GMP by cyclic nucleotide phosphodiesterases. If the effects of SNP were mediated by an increased cGMP level, then the inhibitory effect of SNP on Icap would be expected to be augmented in the presence of a phosphodiesterase inhibitor. Indeed, SNP (100 µM) reduced Icap by 54.7±1.5% (p<0.01, n=9), and this inhibitory effect was augmented by pretreatment with IBMX (1 mM; p<0.01, n=9; Fig. 3B, E).
Next, we examined whether the inhibitory effect of SNP on Icap was mediated by an increased intracellular cGMP concentration. Both 8-bromo-cGMP (100 µM) and 8-pCPT-cGMP (100 µM), which are membrane permeable cGMP analogs, mimicked the inhibitory effect of SNP, decreasing Icap, to 57.1±1.0% (p<0.01, n=5; Fig. 3C, F) and 40.5±1.2% (p<0.01, n=8; Fig. 3C, F), respectively, of the control capsaicin response. The inhibitory effect of 8Br-cGMP co-applied with capsaicin (46.3±2.9% of the control) was not significantly different from that of 8Br-cGMP pretreatment for 1 min before capsaicin application (42.9±8.0% of the control, n=4, data not shown). In addition to these membrane-permeable cGMP analogs, an NO-independent guanylyl cyclase activator, YC-1, was added to the bath to confirm the effect of cGMP on Icap. YC-1 (30 µM) decreased the amplitude of Icap significantly to 41.1±7.1% of the control (p<0.01, n=4; Fig. 3D, F). These results are consistent with the view that the effects of SNP on Icap are mediated through activation of sGC and a resulting increase in intracellular cGMP levels.
A protein kinase G inhibitor abolishes the inhibitory effect of SNP on Icap
To determine whether PKG activation was responsible for the inhibition of Icap, the PKG inhibitor KT5823 (1 µM) was used for 1 min before the co-application of capsaicin and SNP (Fig. 4). The inhibitory effect of SNP on Icap was blocked in the presence of KT5823. We also tested the possibility that protein kinase A (PKA) and protein kinase C (PKC) were involved in the SNP-mediated inhibition of Icap using the PKA inhibitor KT5720 and the PKC inhibitor BIM. Pretreatment with KT5720 (1 µM) or BIM (10 µM) was followed by co-application of capsaicin and SNP. Neither KT5720 nor BIM significantly altered the magnitude of SNP inhibition. The degrees of Icap blockade were 50.2±2.5% (n=6; Fig. 4B) and 29.9±7.1 (n=5; Fig. 4B), respectively, which were similar to the inhibitory effect of SNP alone. These results suggest that PKG may be involved in SNP-induced inhibition of Icap in DRG neurons.
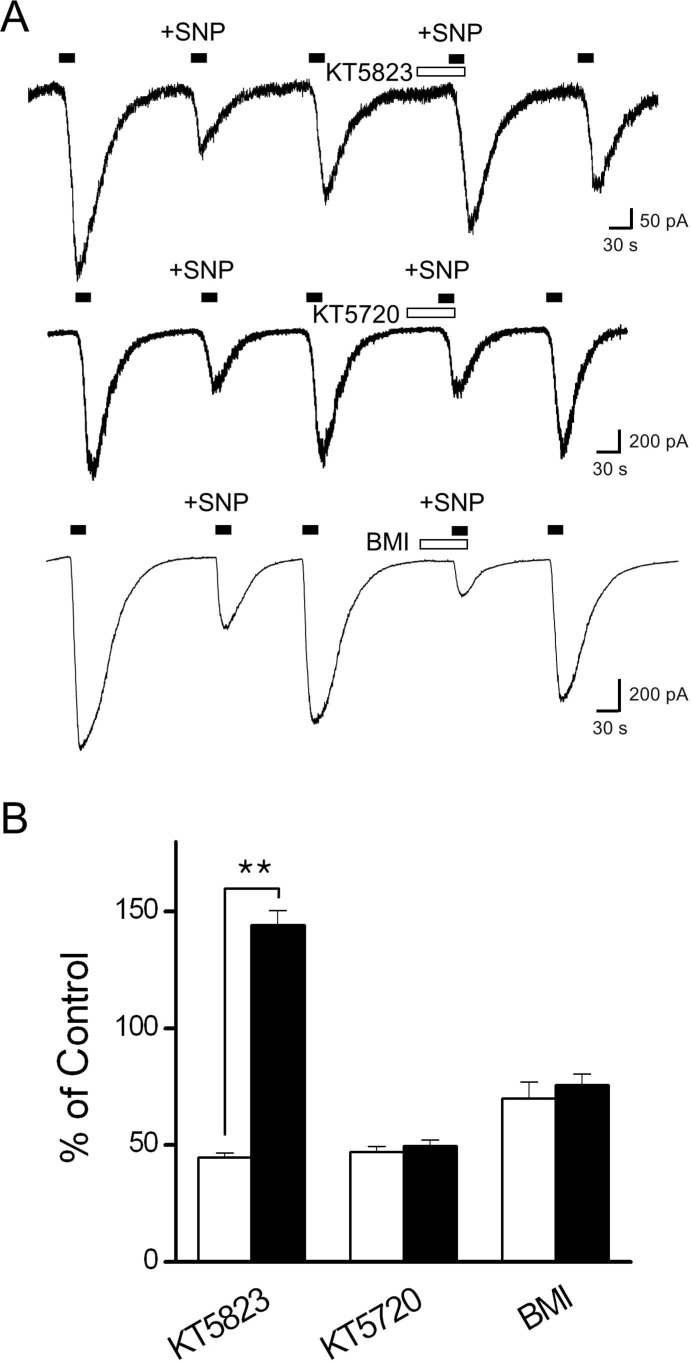 | Fig. 4Effect of the protein kinase G inhibitor, KT5823 on the inhibitory effect of SNP on Icap. (A) KT5823 was applied for 1 min before SNP application. The representative trace shows Icap during the control situation, the application of 100 µM SNP, and the application of SNP in the presence of 1 µM KT5823. Pretreatment with KT5823 potently blocked SNP-induced Icap inhibition and even increased the amplitude of Icap (n=7). Each representative trace shows the effect of SNP in the absence and presence of KT5720 or BIM (10 µM). (B) The bar graph summarizes the effects of SNP on the Icap in the presence (filled bar) and absence (open bar) of the kinase inhibitors. Data are presented as means±SEM. Asterisks indicate significant differences at **p<0.01.
|
Exclusion of TRPV1 S-nitrosylation by 100 µM SNP
To confirm whether NO could activate TRPV1 expressed in DRG neurons by direct S-nitrosylation, we applied SNP (100 µM) or SNAP (100 µM) to the bath for more than 3 min. Neither SNP nor SNAP evoked any membrane current at -60 mV in cells that were sensitive to capsaicin (Fig. 5A). Dithiothreitol (DTT), a sulfhydryl reducing agent, can protect free cysteinyl thiol groups from S-nitrosylation by NO. Thus, we tested whether DTT (10 mM) could prevent the effect of SNP (100 µM) on Icap at the concentration that had prevented sulfhydryl oxidation of TRPV1 [12]. DTT pretreatment did not alter the effect of SNP on Icap (Fig. 5B). These results suggest that 100 µM SNP did not activate or modulate TRPV1 through cysteinyl S-nitrosylation.
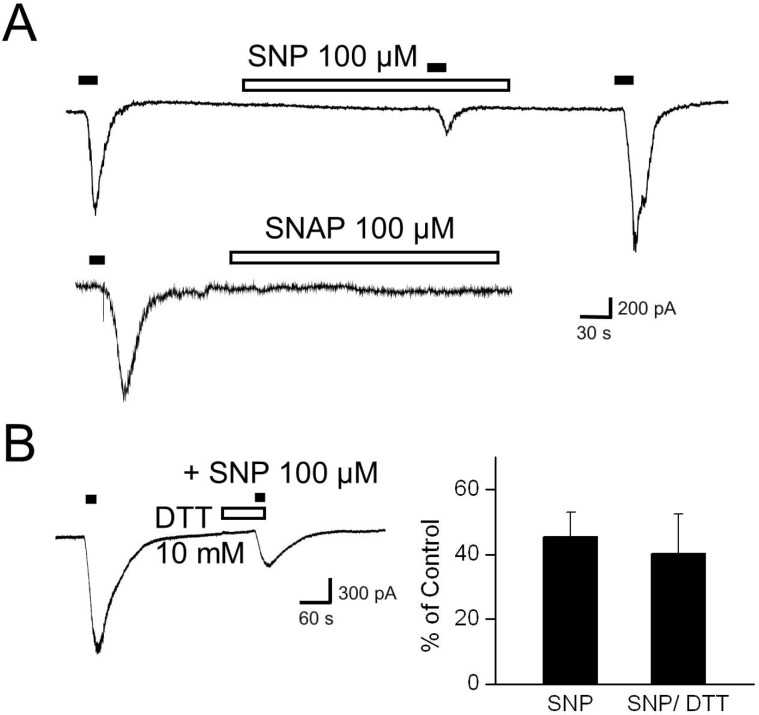 | Fig. 5
S-nitrosylation of TRPV1 by 100 µM SNP. (A) The upper trace shows that SNP (100 µM) alone did not evoke a membrane current at -60 mV in DRG neurons. The lower trace shows that SNAP (100 µM) itself also failed to change the holding current. (B) Dithiothreitol (DTT, 10 mM) was pretreated for 1 min before application of SNP and capsaicin to reduce free sulhydryl groups on TRPV1. The representative trace and bar graph show the effect of SNP in the presence of DTT (n=4).
|
Go to :
DISCUSSION
In this study, we investigated the effects of NO donors on capsaicin-activated whole-cell currents evoked by TRPV1 activation in cultured DRG neurons. Our results demonstrate that NO can modulate TRPV1 indirectly, via activation of the cGMP/PKG-dependent pathway in peripheral sensory neurons.
We investigated the effects of NO donors on capsaicin-activated whole-cell currents in cultured DRG neurons. Our results demonstrate that activation of the cGMP/PKG-dependent pathway by NO can decrease TRPV1 activity in peripheral sensory neurons. We reached this conclusion from the following results. Exposure of DRG neurons to the NO donors SNP and SNAP decreased Icap. In addition, NO scavengers blocked the effects of NO donors. Membrane-permeable cGMP analogs and the sGC activator YC-1 mimicked the inhibitory effect of SNP on Icap. Application of the sGC inhibitor ODQ and the PKG inhibitor KT5823 blocked SNP-induced Icap inhibition.
It has been previously reported that NO can directly activate TRPV1 by S-nitrosylation [28,29]. However, NO has been reported to modulate various ion channels directly, by S-nitrosylation of the channel protein [18-21], or indirectly, by activation of sGC and PKGs [23-25,27]. Additionally, a dual mechanism whereby NO could modulate ion channels by direct and indirect pathways has been described previously in non-neuronal [36,37] and neuronal cells [31,33]. Thus, we could not exclude the possibility that NO could modulate TRPV1 activity via an indirect sGC/PKG pathway. Here, we report that activation of the sGC/PKG pathway by NO can reduce TRPV1 activity in a reversible and concentration-dependent manner in DRG neurons. TRPV1 may be added to the list of ion channels that are modulated by NO via both direct and indirect pathways. Further studies are needed to determine which pathway is most relevant under physiological and pathophysiological conditions.
Miyamoto et al. [28] showed that SNAP activated TRPV1 more potently at positive rather than negative potentials in a cell-attached patch configuration. In contrast, SNAP increased TRPV1 activity throughout the test potentials (voltage ramp from -50 mV to +100 mV) applied to inside-out patches [28]. This suggests that some factors contained in the cytosol may interfere with the activation of TRPV1 by SNAP at negative membrane potentials. It is possible that this interference was mediated by the activation of an indirect sGC/PKG pathway by NO. Although further studies will be needed, membrane-permeable NO seems to activate both nitrosylation of TRPV1 and activation of sGC/PKG simultaneously. Activation of TRPV1 by NO donors may therefore underlie the SNP-induced Icap inhibition in DRG neurons. According to Miyamoto et al. [28], SNAP below 1 mM did not activate TRPV1, but 3 mM SNAP could. Thus, it seems reasonable that we failed to record whole-cell membrane currents induced by relatively low dose of SNAP and SNP at -60 mV (Fig. 5A). Although 100 µM SNP could not open TRPV1 directly in DRG neurons, it was still possible that S-nitrosylation modulated Icap. Assuming that 100 µM SNP could activate both the direct and indirect pathways simultaneously, blockade of either pathway would be expected to affect the magnitude of SNP-induced effects on Icap. That is, preoccupation of free thiol groups of cysteine residues on TRPV1 would leave only the PKG-dependent indirect effects by removing the possibility of direct S-nitrosylation. In contrast, inhibition of PKG could reveal the possible direct effects of SNP. However, the percentile inhibitions of Icap by SNP were not changed significantly in the presence of DTT (Fig. 5B) or the PKG inhibitor (Fig. 4). Thus, we suggest that only the PKG-dependent indirect pathway was activated by NO donors under these experiment conditions using low a concentration (100 µM) of SNP.
Activation of PKG by NO/cGMP can lead to either direct phosphorylation of TRPV1 or stimulation of other proteins, such as a protein phosphatase that dephosphorylates the channel protein [26,38]. Although we observed that the protein phosphatase could be one of the downstream effectors of PKG in the NO effect on TRPV1 (data not shown), TRPV1 itself may be phosphorylated by activated PKG. Thus, further studies will be needed to clarify the target molecule(s) of PKG in the indirect modulation of TRPV1 by NO.
NO has been thought to play important roles in nociceptive signaling. However, the effects of NO in pain transmission are controversial. Morphological data demonstrating nNOS-LI from neurons in nociceptive pathways [39-41] suggest that NO and nociceptive processes are closely associated. However, most experiments using NOS inhibitors have revealed anti-nociceptive effects in various pain models [42-48], and several reports have suggested that peripheral analgesia and blockade of neurogenic edema were related to NO [49-55]. It has been reported that TRPV1 plays a key role in the transmission of thermal pain during inflammation. Thus, dual modulation of TRPV1 by NO may explain the controversy regarding the effects of NO on pain transmission. We suggest that direct activation of TRPV1, through S-nitrosylation, especially at the spinal cord level, may underlie the nociceptive effect of NO. Additionally, NO may inhibit inflammatory thermal pain transmission in neurons, where the downregulation of Icap through PKG activation is the dominant effect of NO.
In the present study, we demonstrated that activation of the indirect cGMP/PKG pathway can inhibit TRPV1 activity in DRG neurons, which may help in understanding the mechanism underlying the peripheral analgesic effect of NO.
Go to :
 XML Download
XML Download