

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Cancer of the oral cavity is an aggressive disease with a high mortality rate when arising in the lip, tongue, floor of the mouth, gingivae, palate, buccal mucosa/vestibule and salivary glands [1,2]. It is generally believed that ethanol (alcohol or ethyl alcohol) and tobacco are the main risk factors in the development of the oral cancer [3,4]. However, despite the potential association between ethanol and oral cancer, no evidence exists to convince ethanol as a promoter of tumorigenesis and ethanol itself is not mutagenic or clastogenic [3,5]. Diverse cellular effects of ethanol in oral cavity region have been reported. Chronic exposure of the oral mucosa to ethanol increased the penetration of carcinogens across the oral mucosa either by enhancing their solubility or the permeability of the oral mucosa [6-9]. Morphological changes in the oral mucosa and the decrease in basal cell size of the esophageal mucosa were observed after exposure to ethanol [10]. In addition, ethanol induced cellular death via apoptosis in certain cell types such as macrophages, human mast cells and the HL-60 promyelocytic leukemia cells [11-13]. Thus, apparently ethanol effects are cell-type dependent and tumor cells are not exceptional. In this context, it is surprising to find that few studies have been undertaken on the effects of ethanol against various tumors originating in oral cavity. Recently, our preliminary study with ethanol showed that tumor cell-types have different sensitivities of cell growth and proliferation to acute ethanol treatment, intriguing us to study further on the ethanol effects in molecular basis. In this study, we investigated the cellular effects of ethanol on YD-15 tongue carcinoma cells, mucoepidermoid carcinoma cells originating in the oral tongue, by exposing cells to various concentrations of ethanol. YD-15 cells showed a high sensitivity to ethanol which was distinct from other cell lines of oral cavity. Further investigation on ethanol effects to YD-15 cells revealed that ethanol elicits inhibitory effect on the growth and proliferation of YD-15 tongue carcinoma cells by mediating cell cycle arrest at low concentration range and induces the cellular death via apoptosis at high concentration.
METHODS
Chemicals
The grade of ethanol used in this study was 99.9% purity and was purchased from Burdick & Jackson (Burdick & Jackson, Muskegon, MI). The ethanol concentration was determined as the percentage volume in culture medium (v/v). Antibodies were purchased from the following sources: p21, cleaved form of caspase-7 (Cell Signaling, Danvers, MA); cdk1, cdk4, cyclin A, cyclin B1, Bcl-2, Bad and Bax and all secondary antibodies (Santa Cruz Biotechnology, Santa Cruz, CA); cdk2, cyclin E1 and PARPp85 (Epitomics, Burlingame, CA). Lysis buffer (1 x solution: 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1 mM Na2EDTA, 1 mM EGTA, 1% Triton, 2.5 mM sodium pyrophosphate, 1 mM β-glycerophosphate, 1 mM Na3VO4, 1 µg/ml leupeptin) was obtained from Cell Signaling. All other chemicals were purchased from Sigma (St Louis, MO).
Cell preparation
YD-15 cells were purchased from the Korean cell line bank (KCLB No 60504, Seoul, Korea) and maintained in RPMI 1,640 media which was supplemented with 10% FBS and antibiotics in a 37℃ incubator at 5% CO2 [14]. Other cell lines used in this study include YD-38 (gingival carcinoma), KB (mouth epidermal carcinoma), FaDu (pharyngeal carcinoma), MCF-7 (breast cancer) and HeLa (cervical cancer). All cell lines were obtained from the Korean cell line bank.
MTT assay
To investigate the effects of ethanol on different human cancer cell lines, cells (4×104/well) in 96-well plates were treated with various concentrations of ethanol and incubated for 24 h. The cells were placed in medium containing 0.5 mg/ml of MTT-1 (Sigma, St Louis, MO). After further incubating for 3 h, media containing MTT-1 were replaced with MTT-2 solution (10% SDS in 0.1 N HCl) by adding 200 µl into each well, and the plates were further incubated for 2 h. The UV absorbance of each sample was then measured at 540 nm. The data were analyzed by Anova single factor. The negative control was the ethanol untreated and used for the determination of percent of live cells.
Cell growth and proliferation assay
Cells in growth phase were transferred into 24-well plates with 3×104 cells/well. After incubation for 24 h, cells were treated with various quantities of ethanol (0, 0.25, 0.5, 0.75, 1.0 and 1.5%) in the culture medium. Cell growth and proliferation were evaluated by an MTT assay and medium replacements contained the same concentrations of ethanol. The OD value of the initial measurement was used as the control.
Western blotting
Cells were treated with various concentrations of ethanol for 24 h and then lysed in a buffer containing freshly added PMSF (1 mM). The cell lysate was homogenized by pipetting for 15 minutes on ice and sonicated for 1 minute. The supernatant of each lysate was collected into new tubes after centrifugation at 20,000 g for 15 minutes at 4℃ and the total protein concentration was measured using a BCA kit (Pierce, Rockford, IL). Antibodies were used in accordance with the manufacturer's instructions. The protein bands were normalized to the beta actin expression level using the Bio-Rad Image Master Program (Bio-Rad, Hercules, CA).
Caspase activity assay
Activated caspase-3 and -7 were detected by using Caspase-Glo® 3/7 Assay kit (Promega, Madison, WI). Cells in 24-well plate (104 cells/well) were exposed to 0, 1.5 or 2.5% of ethanol in medium for 24 h. After ethanol treatment, cells were harvested by using Reporter Lysis buffer (Promega, Madison, WI). Cell lysate was centrifuged at 20,000 g and the supernatant was used for the detection of caspase activities in duplicate. Total protein concentration was measured by using BCA assay kit. Activities of caspases in 1 µg of total protein were normalized with the one of control samples.
DNA content analysis by Fluorescence Activated Cell Sorting (FACS)
Cells were treated with 1.5% or 2.5% ethanol in medium for 24 h. The cells were then harvested by trypsinization and centrifugation at 300 g. After washing of the cell pellets with cold PBS, the cells were fixed in 70% ethanol for 2 h at -20℃, washed again in ice cold PBS and stained with PI solution (20 µg/ml propidium iodide, 200 µg/ml DNase-free RNase-A in PBS) for 15 minutes at 37℃. DNA contents were analyzed by using a Beckman Coulter system.
RESULTS
Comparison of the effects of ethanol on different cell types within the oral cavity
The effects of ethanol on epithelial cancer cells were investigated using cell types originating from different regions of the oral cavity and also in HeLa and MCF7 cells. Oral carcinoma cell lines such as YD-15 (tongue carcinoma), YD-38 (gingival carcinoma), KB cell (mouth epidermal carcinoma), and FaDu (pharyngeal carcinoma) were selected and the effects of ethanol on cell viability were analyzed. YD-15 cells showed the highest growth sensitivity to acute ethanol treatment among this panel (Fig. 1) and the growth inhibition of these cells was even stronger.
Ethanol effects on YD-15 cell proliferation
The effects of ethanol on proliferation were investigated by treating YD-15 cells for 24, 48, 72, 96 and 120 h with various quantities of ethanol (0, 0.25, 0.5, 0.75, 1.0 and 1.5%) in the culture medium. Ethanol was found to inhibit YD-15 cell growth in both a dose- and time-dependent manner (Fig. 2A). A substantial reduction in YD-15 cell survival was observed at ethanol concentrations of above 0.75% but no effects were evident at exposures to less than 0.5% ethanol. In contrast, FaDu squamous carcinoma cells were unaffected by the same alcohol concentrations (Fig. 2B).
Ethanol effects on cell cycle protein inhibitors
To further understand the molecular basis of ethanol effects on cell cycle control, we investigated the expression of cell cycle inhibitors at the lower percent range of ethanol, the condition that ethanol affects on cell cycle without inducing cell death, by treating YD-15 cells with a series of ethanol concentrations for 24 h. The cell cycle inhibitor proteins, p21 and p27, were found to be induced in a dose dependent manner (Fig. 3) and significantly enhanced as the ethanol concentration increased from 0% to 1.5%. The largest induction of p27 and p21 in YD-15 cells was observed at 0.75% and 1.0% ethanol exposures, respectively.
Ethanol effects on cell cycle regulators
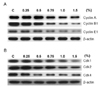
Since cell division is strictly regulated by cell cycle regulators, we further investigated the effects of ethanol on cell cycle regulators such as cyclins and cyclin-dependent protein kinases. The cells were treated with various doses of ethanol (0, 0.25, 0.5, 0.75, 1.0 and 1.5%) for 24 h and lysated for Western blotting. Cylin A, Cyclin E1 and Cyclin B1 were suppressed as the dose of ethanol in the medium increased (Fig. 4A). Band intensities of Cyclin A, Cyclin E1 and Cyclin B1 were gradually decreased in the concentration range of ethanol up to 1.5%. Similarly, ethanol treatment suppressed the expression of cdk-1, -2 and -4 (Fig. 4B). The band intensities of cdk-1 and cdk-2 were gradually decreased as ethanol concentration in the medium increased from 0.25% to 1.5%. Interestingly, the band intensity of cdk-4 was rapidly decreased up to 0.5% ethanol exposure and remained without further decrease at higher ethanol concentrations. This may indicate that cdk-4 is more sensitive to ethanol than cdk-1 or -2, and associated with cell cycle inhibition at an early stage.
Flow cytometry analysis
To further evaluate the nature of the cell cycle arrest caused by ethanol, FACS analysis was carried out. As shown in Fig. 5, the ethanol-exposed YD-15 cells for 24 h resulted in the accumulation of cells at the G2/M phases. Cells treated with a 1.5% dose of ethanol showed a marginal increase in cell distribution at G1/S phase (61.4%) compared to the ethanol untreated cells (56.4%). In contrast, moderate increase, 22.9% to 31.1%, in the cell distribution of ethanol-treated cells at G2/M phase was observed with simultaneous decrease in S phase.
Ethanol effects on the apoptotic response
To determine whether the cell loss caused by ethanol treatment was due to apoptosis or not, we further analyzed the expression of apoptosis related proteins (Bcl-2, Bad and Bax) by western blot, after exposing cells to ethanol at low concentration range of ethanol for 24 h. Ethanol effects on apoptosis associated proteins were minimal at the ethanol range of less than 1.5%. Ratios of Bax/Bcl-2 and Bad/Bcl-2 were relatively constant and the cleaved form of PARP (p85), a major hydrolysis product of executor caspases, was only weakly detected, suggesting that ethanol did not affect cell death at this range of ethanol concentration (Fig. 6A). However, at 2.5% ethanol, ethanol induced YD-15 cell death via apoptosis. Strong bands of the cleaved form of PARP (p85) and the activated form of caspase 7 (20 kDa) were detected in Western blotting (Fig. 6B). In addition, dramatic increase of caspase-3 and -7 activities were detected in caspase activity assay (Fig. 6C). The results indicate that ethanol suppressed cell cycle progress in the low range of ethanol concentration without inducing apoptosis.
DISCUSSION
In this study we investigated the cellular effects of ethanol on YD-15 tongue carcinoma cells [15] by exposing cells to various concentrations of ethanol. YD-15 cells showed a high sensitivity to ethanol which was clearly distinguished from other cell lines of oral cavity (Fig. 1, 2). Further investigation on ethanol effects to YD-15 cells revealed that ethanol elicits inhibitory effect on the growth and proliferation of YD-15 tongue carcinoma cells by mediating cell cycle arrest at G2/M at low concentration range and induces the cellular death via apoptosis at high concentration. The inhibitory ethanol effects on the growth and proliferation of YD-15 cells was confirmed by altered expression of both cell cycle inhibitors and cell cycle regulators. P27 and p21 cell cycle inhibitors were up-regulated dose-dependently by ethanol (Fig. 3) whilst the expression of cell cycle regulators, cdks and cyclins, was down-regulated (Fig. 4). P27 blocks G1 progression by binding and inactivating the cyclin E/cdk2 and cyclin D/cdk4 complexes [16-18] and p21 is also known to inhibit a broad range of the cell cycle proteins, including G1 cyclin/cdk complexes and cyclin B1/cdk1 complexes [19-22]. Thus, the up-regulation of p27 and p21 by ethanol in YD-15 cells may cause the reduced expression of cdk2 and cdk4. In addition, the expression of both cyclin B1 and cdk1 was suppressed by ethanol treatment, limiting the supply of the cdk1/cyclin B complex required for the G2 to M phase transition and thereby delaying the phase transition. FACS analysis further confirmed the cell cycle arrest at G2/M phase in cells exposed with 1.5% ethanol (Fig. 5). Despite the suppression of cell cycle arrest at a low percentage of ethanol, ethanol did not promote the apoptosis of YD-15 cells, suggesting that the cell cycle inhibition by ethanol was not sufficient to induce apoptosis at a low percentage of ethanol (less than 1.5%) (Fig. 6). Conversely, YD-15 cells exposed to a high percentage of ethanol (2.5%) for 24 h induced significant programmed cell death with the appearance of the cleaved PARP (the marker of caspase-3 mediated apoptosis) and the activated form of caspase-3 and -7.
The lateral border of the tongue, one of the non-keratinized tissues, is known to be more permeable than keratinized tissues such as the palate and gingivae [23]. Thus the unusual sensitivity of YD-15 cells to ethanol could be due to the enhanced cellular uptake of ethanol by the increased lipid membrane permeability. The fast passage cycle of tongue carcinoma cells may also contribute to the ethanol sensitivity of YD-15 cells. Interestingly, previous reports also showed the inhibitory effect by ethanol on the proliferation of cancer or non-cancer cells such as fibroblast growth factor 1-induced human smooth muscle cells and smooth muscle cell in the postprandial state [11,24-28]. In addition, a recent study by Clemens et al demonstrated that ethanol treatment in recombinant Hep-G2 cancer cells caused a G2/M cell-cycle arrest [29]. To our best knowledge, this is the first report that ethanol specifically suppresses the growth and proliferation of tongue carcinoma cells among various tumor cell-types originating in oral cavity. The question how ethanol induces the cell cycle arrest and what is the signal transition mechanism underlying the cell cycle arrests remain to be answered in future study. Overall, our results suggest that the cellular inhibitory effects by ethanol may function as a tumor suppressor in the oral tongue cancer.





 XML Download
XML Download