

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Tautomycetin (TMC) is classified as a type I PKS-derived compound which was first isolated in 1989 from Streptomyces griseochromogenes, a gram-positive mycelia bacterium [1]. It was originally described as an antifungal antibiotics. However, TMC was later found to be able to specifically inhibit protein phosphatase type 1 (PP1) and protein phosphatase type 2A (PP2A) [2]. In contrast to other naturally occurring PP1 and PP2A inhibitors, such as okadaic acid, calyculin-A, fostriecin, and cantharidin, TMC exhibits a high degree of PP1 selectivity [3-5]. TMC preferentially inhibits PP1 by a nearly 40-fold relative to PP2A and is the most selective PP1 inhibitor reported to date [2]. Furthermore, since TMC is a linear aliphatic inhibitor of PP1, and not a cyclic peptide like microcystins and nodularins [6,7], it is an attractive lead compound for novel PP1-specific drug design.
The protein phosphatase PP1 is tightly regulated by its interaction with more than 180 known targeting proteins which localize PP1 and its inhibitory proteins to distinct regions of the cell [8,9]. The broad spectrum of PP1 binding partners is consistent with the wide distribution of PP1 throughout the human body and its multiple regulatory roles in cellular processes such as cell-cycle progression, protein synthesis, muscle contraction, carbohydrate metabolism, transcription and neuronal signaling. Given the importance of the interplay between phosphatases and kinases in human diseases, the selective inhibition of PP1 and PP2A has been proposed to be an attractive goal for rational anticancer drug design [10].
TMC represents not only an interesting drug lead but also a powerful biochemical tool to elucidate the roles of PP1 in various biological pathways. TMC has been suggested as a potential drug for colorectal cancer chemotherapy because of its regulation of Raf-1 activity in a colon cancer cell-type-specific manner. The anti-proliferation effect of TMC on human colon cancer cell lines is found to be accompanied by induction of the level of cell cycle regulator p21Cip/WAF1 through the Raf/ERK pathway. Although TMC treatment led to activation of the Raf-1-MEK-ERK cascade, the level of GTP-bound Ras was not altered, indicating that TMC may mediate its effects through regulation of Raf-1 activity [11].
TMC has been also identified as a potent immunosuppressor of activated T cells both in vitro and in vivo [12]. Inhibition of T cell proliferation by TMC was observed at concentrations 100-fold lower than those needed to achieve maximal inhibition by cyclosporine A (CsA). CsA and FK506 exert their pharmacological effects by binding to the immunophilins; the resulting complex binds to and inhibits the Ser/Thr phosphatase calcineurin albeit with potentially deleterious effects due to the physiological ubiquity of calcineurin [13,14]. TMC exerts immunosuppressive activity in a manner completely different from those of CsA and FK506 by blocking tyrosine phosphorylation of T-cell-specific signaling mediators. This leads to cell-specific apoptosis due to cleavage of Bcl-2, caspase-9, caspase-3, and poly (ADPribose) polymerase. The activated T cell specificity of TMC thus suggests this unique polyketide as a significant lead in the search for immunosuppressive drugs superior to CsA and FK506 [12].
The immunosuppressive and anticancer activities of TMC are both thought to be related to its ability to inhibit protein phosphatases. Unfortunately, this hypothesis has not yet been fully clarified. On the other hand, it is of importance to gain more insights into its structure-activity relationship due to the increasing use of TMC as a biological probe in molecular and cellular biology. In the present study, the biological activities of TMC and its derivatives were examined, and the interrelationship between the biological activities and the chemical structures was discussed.
METHODS
Generation of TMC mutant strains
The tauF, tauD and tauK genes were inactivated using a PCR-targeted gene-disruption system according to the literature protocols [15]. Briefly, an apramycin resistance gene aac(3)IV/oriT cassette was used to replace an internal region of each target gene. Mutant cosmids pTAUTF (ΔtauF), pTAUTD (ΔtauD) and pTAUTK (ΔtauK) were constructed and introduced into S. griseochromogenes by conjugation from E. coli ET12567/pUZ8002 according to the literature procedure illuminated the part of conjugation procedure above. The desired double crossover mutants, selected by the apramycin-resistant and kanamycin-sensitive phenotype, were isolated as STQF (ΔtauF), STQD (ΔtauD) and STQK (ΔtauK).
Extraction of TMC and its derivatives
TMC and TMC-OH were isolated from the fermentation broth of wild strain, while TMC-F1, TMC-D1, TMC-D2, TMC-K1 and TMC-K2 were isolated from the fermentation broth of ΔtauF, ΔtauD, ΔtauK mutant strains respectively. TMC-Ring and TMC-Chain were obtained by chemical degradation of TMC with CsCO3. All these compounds were analyzed and purified by HPLC with CH3CN / H2O (0.5% acetic acid) as mobile phase. Compound identity was confirmed by high-resolution mass spectrometry and no impurities were detected by 1H-NMR.
Animals and cell culture
Female Balb/c mice between 8 and 14 weeks of age were housed under SPF conditions. The experimental procedure was reviewed and approved by the Dalian University of Technology Animal Research Committee. The Jurkat T cell, MCF-7 human breast cancer cell line and HCT-15 human colon cancer cell line were maintained in the RPMI 1640 medium containing 10% fetal bovine serum, antibiotics (100 U penicillin/ml and 100 µg streptomycin/ml), and 1.5 mM L-glutamine at 37℃ in a 5% CO2 humidified incubator.
Cell proliferation assay
MCF-7 and HCT-15 tumor cell proliferation was measured by 3-(4,5-dimethylthiazol-2-yl)-2,5 diphenyl tetrazolium bromide (MTT) rapid colorimetric assay [16]. Briefly, cells were seeded in quadruplicate on 96-well plates and incubated overnight under standard conditions to allow cell attachment. The cells were then treated with TMC and its derivatives at concentrations of 0 to 100 µM and incubated for up to 72 hours. The MTT assay was performed by replacing the standard medium with 100 µl PBS containing 0.5 mg/ml MTT and incubating at 37℃ for 4 hours. After incubation, the crystals were dissolved with 200 µl dimethyl sulfoxide. The multiwell plates were then measured at 540 nm using a spectrophotometer (Molecular Devices).
Assay of PP1 phosphatase activity
The activities of PP1 was assayed with 6,8-difluoro-4-methylumbelliferyl phosphate (DiFMUP) as a substrate (EnzChek phosphatase assay kit, Invitrogen, Madison, WI). Briefly, recombinant PP1 (NEB) was diluted with reaction buffer (50 mM HEPES, 100 mM NaCl, 1 mM MnCl2, 2 mM DTT, 0.01% Brij-35), then preincubated with TMC or its derivatives for 20 min at 30℃. The substrate for PP1 was added and samples were incubated at 30℃ in a shaking incubator for 30 min. Then phosphatase activity was measured in a fluorometer using excitation at 360 nm and emission detection at 460 nm. For in vivo PP1 phosphatase assays, MCF-7 cells were grown to subconfluence in 100-mm dishes with or without TMC or its derivatives for 4 h at 37℃. Thereafter, growth media were removed from control or drug-treated cultures, which were then washed twice with phosphate-buffered saline. Following this, 0.2 ml of lysis buffer (20 mmol imidazole-HCl, 2 mmol EDTA, 2 mmol EGTA with 10 µg/ml each of aprotinin, leupeptin, pepstatin, 1 mmol benzamidine, and 1 mmol phenylmethylsulfonyl fluoride) was added to each dish. Cells were scraped into a plastic tube and centrifuged at 10,000 g and 4℃ for 10 min to give a soluble supernatant. Lysates were incubated with an additional 2 nM or 100 nM okadaic acid for 15 min before the labeled substrates were added. PP1 activity is defined as the activity insensitive to 2 nM okadaic acid but inhibited by 100 nM okadaic acid. Subsequently, the supernatants were assayed for PP1 activities using a phosphatase assay kit as described above for the measurement of recombinant PP1 activity [17].
T cell proliferation assay and IL-2 ELISA
Primary T cells were isolated from the spleen of mice. Two spleens were converted into single-cell suspensions by squeezing through a cell strainer with the rough end of a 3-ml syringe plunger. Cells were spun down in a 50-ml Falcon tube (5 min, 300 ×g) and the supernatant was discarded. The pellet was vigorously resuspended in erythrocyte lysis buffer (5 ml/spleen) and then incubated for 5 min at room temperature. The mixture was filled up with PBS to 30 ml, spun down (5 min, 300 ×g) and resuspended in 5 ml PBS. Sequentially, CFSE was added at 10 µm/ml for labeling at room temperature for 5 min, followed by adding 25 ml RPMI containing 10% FCS, spin at same as above. CFSE used for labeling of the CD4 and CD8 T cells to monitor the proliferation. Cells were counted and resuspended at 5×106 per ml containing 50 U/ml human IL-2. Next, the T cells were placed into the plates coated with anti-CD3 and -CD28 Ab at 100 µg/ml and 50 µg/ml respectively. The cells were incubated in 48 well-plates for 3 days. The CD4 and CD8 T cells were analyzed by FACS method. The function of compounds will be determined by CFSE dilution of cell contents.
For viability assay of Jurkat T cell, the cells were treated with TMC and its derivatives at concentrations of 0 to 100 µM and incubated for up to 72 hours. Cell viability was determined using a Cell Proliferation Assay kit (Promega, USA). The MTS assay is a colorimetric method that uses a tetrazolium compound 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS), and an electron-coupling reagent, phenazine methosulfate (PMS). The cells chemically reduce MTS into formazan, which is soluble in tissue culture medium. MTS was then added to each well and the plates were read at 490 nm wavelength three hours later using an ELISA reader (Molecular Devices).
Jurkat T cell IL-2 cytokine levels in conditioned medium were determined using a commercial ELISA kit. It detects soluble IL-2 with a monoclonal antibody specific for IL-2. Briefly, the monoclonal antibody will bind IL-2 in cell culture supernatants during incubation. After several washes to remove unbound proteins, an enzyme-linked polyclonal antibody was added that binds to the solid phase bound IL-2. After washing, the substrate solution was added and developing color was measured using a multiwell plate reader set to 450 nm. The optical density of the samples was compared to a calibration curve.
RESULTS
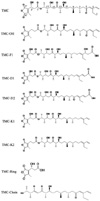
Structure of TMC and its analogous compounds
As the most specific inhibitor of protein phosphatase 1 in vitro, TMC has been reported to have anti-fungi, anti-tumor and immunosuppressive activities [2,12], which make TMC and its analogs excellent agents for potential clinical use as well as for biological studies. To dissect the relationship between protein phosphatase inhibition and other biological functions of TMC, we obtained a series of TMC analog compounds via genetic engineering of the TMC biosynthetic gene cluster, by isolation from fermentation broth, or through chemical degradation. The structures of the compounds used in this study are shown in Fig. 1. Compared with TMC, TMC-OH lacks the hydroxyl group at the C3' position. TMC-F1, TMC-D1 and TMC-D2 all lack the terminal C3 olefin, a critical feature of the right hemisphere of TMC. TMC-F1 and TMC-D2 have β-hydroxypropanoic acid and acrylic acid attached to C3 position respectively. TMC-D1 lacks carbonyl group at the C5 position and has an acrylic acid attached to C3. Both TMC-K1 and TMC-K2 lacks carbonyl group at the C5 position compared to their parent structures TMC and TMC-OH. Lastly, TMC-Ring possesses only the dialkymalic anhydride moiety whereas TMC-Chain possesses only a carbon chain.
Effect of TMC and its related compounds on PP1 activity
We then set to determine the effect of the structural changes on inhibition of PP1 activity. As shown in Fig. 2, TMC and its related compounds inhibited activities of the catalytic subunits of PP1 with a wide range of IC50 values (from 0.5 nM to 40 µM). The magnitude of the inhibitory activity was in the order of TMC > TMC-K1 > TMC-D1 ≈ TMC-F1 ≈ TMC-OH > TMC-D2 > TMC-K2 > TMC-Ring > TMC-Chain. Table 1 shows the IC50 values of all compounds examined. The IC50 values of TMC-K1, TMC-D1, TMC-F1, TMC-OH and TMC-D2 were estimated to be 16 nM, 101 nM, 128 nM, 200 nM and 610 nM respectively. Among all the TMC derivatives, TMC-Chain demonstrates the lowest IC50 values of 32 µM against PP1.
Effects of TMC and its related compounds on anticancer activity
We evaluated the cytotoxicity of TMC and its related compounds against HCT-15 colon adenocarcinoma and MCF-7 breast carcinoma cell lines. The IC50 values of TMC and its related compounds for inhibition of cancer cell growth are summarized in Table 2. TMC, TMC-OH, TMC-D2, TMC-K1 and TMC-Chain inhibited the growth of HCT-15 cells with IC50 values of 0.35 µM, 13 µM, 8 µM, 8 µM and 10 µM respectively. The IC50 values of the rest of the compounds (TMC-F1, TMC-D1, TMC-K2 and TMC-Ring) are all beyond 50 µM. The IC50 values of TMC and its derivatives for inhibiting MCF-7 cells growth were almost the same as those for inhibiting HCT-15 cells growth.
Effects of TMC and its related compounds on immunosuppressive function
TMC exhibits its immunosuppressive activity via T cell-specific apoptosis induction. We evaluated the cytotoxicity of TMC and its related compounds against primary T cells (Table 3). While the analogs TMC-F1, TMC-D1, TMC-K2 and TMC-Ring did not show any inhibitory effect at concentrations below 20 µM, TMC-OH, TMC-K1 and TMC-Chain retained significant, albeit reduced, cytoxicity (within 35-fold of reduction relative to TMC). Cytotoxicity of TMC and its related compounds against Jurkat T cell lines was also evaluated (Table 4). TMC, TMC-OH, TMC-D2, TMC-K1 and TMC-Chain inhibited the growth of Jurkat T cells with IC50 values of 0.28 µM, 18.2 µM, 10 µM, 5.4 µM and 9.6 µM respectively. The IC50 values of the rest of the compounds (TMC-F1, TMC-D1, TMC-K2 and TMC-Ring) are beyond 50 µM. Remarkably, production of IL-2 was almost completely suppressed (98%) by TMC (200 nM), TMC-OH, TMC-D2, TMC-K1 and TMC-Chain (20 µM). In contrast, TMC-F1, TMC-D1, TMC-K2 and TMC-Ring did not show significant inhibition of IL-2 production in the same range of concentrations (Fig. 3).
Cytotoxicity of TMC doesn't correlate with its in vivo PP1 inhibition activity
To further investigate the relationship between cytotoxicity of TMC and its PP1 inhibitory activity, we measured in vivo phosphatase activity of PP1 in MCF-7 cell extracts treated with TMC and its related compounds (Fig. 4A). TMC (1 and 2 µM) decreased PP1 activities by 60% and 81% compared to untreated cells, respectively. The cell growth exposed to 1 and 2 µM TMC were inhibited by 90% and 96% relative to untreated cells (Fig. 4B). The PP1 activities treated with 20 and 50 µM TMC-F1 were also reduced by 38% and 60%, respectively. In contrast, TMC-F1 did not significantly affect the growth of MCF-7 cells with the inhibition ratio of 8% and 24%, respectively. In vivo activities of PP1 in extracts prepared from TMC-Chain (20 and 50 µM)-treated cells were only reduced by 9% and 19% respectively. However, TMC-Chain significantly affected the growth of MCF-7 cells, with a decrease of 70% and 89% respectively. These results indicate that the cytotoxicity of TMC doesn't correlate with its in vivo PP1 inhibitory activity.
DISCUSSION
We have studied the relationship between the chemical structure and the function of TMC and its derivatives. Our results could provide new insight into the relationship of protein phosphatase inhibition with cell killing capacity.
To investigate the impact of right hemisphere modification upon TMC bioactivity, TMC related compounds were subjected to PP1 inhibition and cytotoxicity assays. The results showed that immunosuppressive activity was accordant with anticancer activity for TMC and its analog compounds. For instance, the IC50 values for inhibiting MCF-7 cell growth are accordant with inhibiting Jurkat T cell growth for all TMC derivatives. These results indicate that TMC may home at the same targets for its immunosuppressive and anticancer activity. TMC-F1 and TMC-D1 both retained significant, albeit reduced, PP1 inhibitory activity, while the two compounds did not show immunosuppressive and anticancer activity. TMC-OH showed significant immunosuppressive and anticancer activity, with potent PP1 inhibitory activity similar to TMC-F1 and TMC-D1. TMC-D1 and TMC-D2 are structurally similar, differing only in the presence of a carbonyl group at the C5 position on TMC-D2. TMC-D1 had a more significant PP1 inhibitory activity than that of TMC-D2, whereas it showed less cytotoxicity. Moreover, TMC-Chain did not show any significant inhibitory activity for PP1 but similarly showed strong growth inhibitory effect. This fact demonstrates that the maleic anhydride moiety of TMC is critically important for its phosphatase inhibitory activity but not for its growth inhibitory effect, whereas the C1-C18 moiety of TMC is essential for the inhibition of cell proliferation.
In order to further confirm the relationship between cytotoxicity of TMC and its PP1 inhibitory activity, we measured in vivo phosphatase activities of PP1 in MCF-7 cell. The in vivo PP1 activities treated with TMC-F1 and TMC-Chain (50 µM) were reduced by 60% and 19%, respectively. In contrast, growth of cells exposed to the same concentration of TMC-F1 and TMC-Chain were inhibited by 19% and 89% relative to untreated cells, respectively. However, even if the in vivo PP1 inhibitory activity was almost completely lost, TMC-Chain (20 µM) showed relative potent cytotoxic activity. Moreover, both TMC (2 µM) and TMC-F1 (50 µM) reduced the PP1 activity by 60%, whereas the cell viabilities were inhibited by 90% and 24%, respectively. These results indicate that the cytotoxicity of TMC does not correlate with its in vivo PP1 inhibition activity.
It has been previously reported that thyrsiferyl 23-acetate and tautomycin exhibit apoptosis-inducing activity in addition to their inhibitory effect on protein phosphatase [18,19]. Similarly, our studies also demonstrated that TMC can inhibit cell growth by inducing apoptosis (data not shown) besides its phosphatase inhibitory function. The present work provides further evidence that the phosphatase-inhibitory activity and the growth inhibitory effect could be attributed to different portions of the natural compound. A recent report suggests that SHP2 (Src homology-2 domain containing protein tyrosine phosphatase-2) is a potential target for the immunosuppressive activity of TMC. Specifically, TMC and TMC-D1 inhibit SHP2 with a similar IC50 values whereas TMC-D2 does not exhibit significant inhibitory activity against SHP2 [20]. However, our data demonstrated that TMC-D2, instead of TMC-D1, exhibited significant immunosuppressive activity. This is inconsistent with the proposed target model. If SHP2 is a cellular target for TMC, it can not explain the mechanism for TMC-D2's observed immunosuppressive and anticancer activity. In this regard, we do not rule out the possibility that TMC's biological activities could be attributed to its interaction with other cellular proteins. Thus, some unidentified molecular target may be required to explain the biological activities of TMC.
PP1 are critical signaling molecules that regulate many cellular processes including cell cycle progression, cell growth, gene expression, calcium transport and neuronal signaling [21]. Therefore, one prevailing hypothesis is that the TMC-induced apoptosis occurs through the inhibition of PP1 [11,12]. Remarkably, our study has implicated that different portions of TMC structure play a distinct role in protein phosphatase inhibition and the induction of apoptosis. Our data suggest that the immunosuppressive and anticancer activity of TMC is not only due to the inhibition of PP1. Therefore, we suggest that TMC may have multiple targets for its biological activities. Multiple blockades of these molecular targets may produce additive and even synergistic cytostatic effects. Identification of potential novel targets responsible for different effects of TMC is underway.







 XML Download
XML Download