

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Gout is caused by hyperuricemia through altered purine metabolism. Although asymptomatic hyperuricemia is common and the majority of patients never develop gout [1], gout is a common cause of arthritis affecting more than 1% of the adult population [2]. When the local concentration of uric acids exceeds the limit of solubility, monosodium urate (MSU) crystals are generated and precipitate in the joints, kidneys, and soft tissues, causing inflammation and leading to gout [3]. MSU crystals stimulate local connective tissue cells, monocyte-macrophages, and neutrophils to produce a variety of inflammatory cytokines including tumor necrosis factor (TNF)-α, interleukin (IL)-8, IL-1β, IL-6 and monocyte chemotactic factor, which collectively induce acute inflammation [4,5].
Nitric oxide (NO) is a small gas molecule synthesized by three isoforms of NO synthase (NOS) [6]. Although NO from two constitutive isoforms (endothelial NOS and neuronal NOS) is critical in a wide variety of physiological functions [6], overexpression of inducible NOS (iNOS) is involved in a variety of pathological conditions including inflammation [7,8]. Although the major metabolites of NO are nitrite and nitrate [9], NO can be transformed to peroxynitrite under oxidative stress, which produces nitrated proteins including nitrotyrosine [10]. Nitrotyrosine is a marker of peroxynitrate production and nitrative stress [11,12]. Expression of iNOS is also increased in MSU-stimulated chondrocytes and in the synovial tissue of gouty arthritis patients, suggesting a potential role of iNOS in the pathogenesis of arthritis [13,14]. However, no direct evidence of the involvement of iNOS in gout has been reported. In this study, the role of iNOS in gouty arthritis was elucidated by examining whether a selective iNOS inhibitor suppressed MSU-induced inflammation in a mouse foot model.
METHODS
Animals
Seven-week-old male C57BL/6 mice were housed in a room operating with a 12:12 h light/dark cycle. All the mice were fed a standard chow diet with free access to water. For the preparation of MSU crystals, 4 g of uric acid was dissolved at 60℃ in 800 ml of 0.5 M NaOH (pH 8) and cooled overnight at 4℃. After discarding the supernatant, precipitated crystals were collected, washed and dried. The needle-like crystals were dissolved in saline (0.04 g/500µl) and 4 mg/50µl of the solution was injected into soles of hindlimb feet of the mice [15]. This concentration is high enough to induce inflammation since the solubility of uric acid in plasma is 6.8 mg/dl [16]. The iNOS selective inhibitor N6-(1-iminoethyl)-L-lysine (L-NIL; Cayman Chemical, Ann Arbor, MI, USA)[17,18] was injected into mice intraperitoneally (10 mg/kg) 4 h before MSU injection. Twenty four hours after MSU injection, edema of feet was measured using digital calipers (Mitutoyo Corporation, Kawasaki-shi, Kanagawa, Japan) in mice anesthetized with an intraperitoneal injection of tiletamine and zolezepam (25 mg/kg) and zylazine (10 mg/kg). Blood was collected from the retro-orbital plexus using micro-hematocrit capillary tubes coated with heparin. The blood was centrifuged and plasma was stored at -80℃ for further analysis. The feet were excised and stored at -80℃ for further analysis. This study was conducted in accordance with the guidelines for the care and use of laboratory animals provided by Yeungnam University, and all experimental protocols were approved by the Ethics Committee of Yeungnam University.
Cell culture
Human dermal fibroblast (HDF) was purchase from Lonza (Walkersville, MS, USA) and mouse myoblast cell line, C2C12, and human fetal osteoblastic cell line, hFOB1.19, were purchased from the American Type Culture Collection (Manassas, VA, USA). HDF and C2C12 were culture in Dulbecco's Modified Eagle Media (DMEM; GIBCO, Grand Island, NY, USA) at 37℃ in a 5% CO2-humidifier incubator. hFOB1.19 cells were cultured in a 1:1 mixture of DMEM and F12 (GIBCO) without phenol red at 36.5℃ in an atmosphere containing 5% CO2. The cells were plated at 1×106 to each of 6 wells and treated with 2 mg MSU for 2 h. L-NIL (0.25 or 0.5 mg/ml) was added into the cells 30 min before MSU treatment. RNA extraction was performed for the quantitative real time polymerase chain reaction (qRT-PCR).
qRT-PCR
qRT-PCT was performed as previously described [19]. Briefly, 25 mg tissue samples were homogenized in TRI reagent (Sigma-Aldrich, St. Louis, MO, USA) and RNA was reverse transcribed to cDNA using a reverse transcription kit (Applied Biosystems, Foster City, CA, USA). Quantitative real-time PCR was performed using the Real-Time PCR 7500 System and Power SYBR Green PCR Master Mix (Applied Biosystems) according to the manufacturer's instructions. Expression levels of β-actin were used for sample normalization. Each reaction mixture was incubated at 95℃ for 10 min followed by 45 cycles of 95℃ for 15 s, 55℃ for 20 s and 72℃ for 35 s. Sequences of primers were based on the National Center for Biotechnology Information nucleotide database and were designed using the Primer Express Program (Applied Biosystems). The primer sequences were: mouse β-actin (121 bp: forward, 5'-TGG ACA GTG AGG CAA GGA TAG-3'; reverse, 5'-TAC TGC CCT GGC TCC TAG CA-3'), mouse iNOS (71 bp: forward, 5'-CTC CTG CCT CAT GCC ATT-3'; reverse, 5'-TGT TCC TCT ATT TTT GCC TCT TTA-3'), mouse TNF-α (71 bp: forward, 5'-CTA TCT CCA GGT TCT CTT CAA-3'; reverse, 5'-GCA GAG AGG AGG TTG ACT TTC), mouse IL-1β (71 bp: forward, 5'-GCC CAT CCT CTG TGA CTC-A-3'; reverse, 5'-AGT GCA GCT GTC TAA TGG GA-3'), mouse superoxide dismutase (SOD; 71 bp: forward, 5'-CTG CTC TAA TCA GGA CCC ATT-3'; reverse, 5'-GTG CTC CCA CAC GTC AAT C-3'), mouse glutathione peroxidase 1 (GPx1; 71 bp: forward, 5'-GAA GTG CGA AGT GAA TGG TG-3'; reverse, 5'-TGG GTG TTG GCA AGG C-3'), human β-actin (72 bp: forward, 5'-ACC GCA TCG TCA CCA AC-3'; reverse, 5'-CCA CAC GCA GCT CAT TGT A-3'), human iNOS (192 bp:forward, 5'-TTA TGA CTC CCA AAA GTT TGA CCA-3'; reverse, 5'-CCG TCA GTT GGT AGG TTA CTG TTG-3'), human TNF-α (72 bp: forward, 5'-AGA GGG CCT GTA CCT CAT CTA-3'; reverse, 5'-AGC AGC ACA TGG GTC GAG-3'), and IL-1β (133 bp: forward, 5'-TCC AGG GAC AGG ATA TGG AG-3'; reverse, 5'-TCT TTC AAC ACG CAG GAC AG-3').
Western blotting
Foot samples were used for measurement of the protein level of phosphorylated extracellular signal-related kinase (pERK; Cell Signaling Technologies, Danvers, MA, USA), ERK (Cell Signaling Technologies), p38 (Cell Signaling Technologies), p-p38 (Cell Signaling Technologies), iNOS (Santa Cruz Biotechnology, Santa Cruz, CA, USA), nitrotyrosine (Upstate Biotechnology, Lake Placid, NY, USA), and glyceraldehyde 3-phosphate dehydrogenase (GAPDH; Santa Cruz Biotechnology). Western blotting was performed as previously described [12]. Briefly, 25 mg tissue samples were homogenized in lysis buffer (Invitrogen, Carlsbad, CA, USA) and extracted protein was separated by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE). Resolved proteins were transferred to a polyvinylidene fluoride membrane (Millipore, Billerica, MA, USA). After blocking with 5% skim mile, the membrane was incubated overnight at 4℃ with primary antibody and then specific antibody binding was detected using sheep anti-rabbit IgG horseradish peroxidase or goat anti-mouse IgG horseradish peroxidase (Bio-Rad, Hercules, CA, USA) for 1 h at room temperature, except for nitrotyrosine, which was detected using mouse anti-mouse IgG horseradish peroxidase (Bio-Rad). The binding was visualized using an enhanced chemiluminescence detection regent (Millipore).
Nitrite and nitrate
Plasma concentrations of the NO-derived end products nitrite and nitrate were measured by a TotalNOAssay Kit (R&D Systems, Minneapolis, MN, USA). To minimize interference with plasma protein, the sample was ultra-filtered through a 10 kDa cut-off filter (Millipore) prior to the assay.
RESULTS
Feet edema
MSU injection into the soles induced redness, swelling, and heat in mice feet (data not shown) and significantly increased the thickness of feet by 160% at 24 h compared with the saline injected feet. Pretreatment of 5 and 10 mg/kg L-NIL suppressed MSU-induced edema by 12% and 40%, respectively, while L-NIL alone had no effect on the thickness of feet (Fig. 1). Since 10 mg/kg/day L-NIL significantly reduced MSU-induced foot pad swelling, this concentration of L-NIL was used in this experiment.
iNOS expression
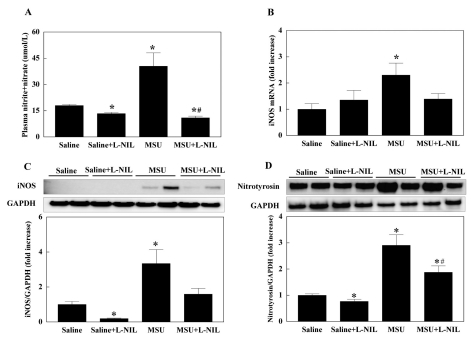
Plasma levels of the NO metabolites nitrite and nitrate was reduced by L-NIL administration compared with control. MSU injection increased the NO metabolites in plasma, which was reversed by L-NIL pretreatment (Fig. 2A). While gene expression of iNOS was not affected by L-NIL, the mRNA level of iNOS in feet was significantly increased by MSU. L-NIL pretreatment suppressed MSU-induced gene expression of iNOS (Fig. 2B). Protein level of iNOS in feet showed the same pattern of changes as with iNOS gene expression, except that L-NIL treatment alone significantly reduced the level of iNOS protein (Fig. 2C). Nitrotyrosine level in feet was significantly increased by MSU injection, which was partially suppressed by L-NIL pretreatment. Like the iNOS protein level, nitrotyrosine was significantly reduced by L-NIL treatment (Fig. 2D).
Gene expression of inflammatory cytokines and antioxidant enzyme
Gene expression of TNF-α was also increased by MSU, which was significantly reduced by L-NIL pretreatment. However, MSU-induced TNF-α expression was not completely normalized by L-NIL pretreatment compared with saline-injected control mice. L-NIL treatment did not alter the mRNA level of TNF-α. While L-NIL had no effect on the gene expression of IL-β, the mRNA level of IL-1β was significantly increased by MSU. L-NIL pretreatment suppressed MSU-induced gene expression of IL-1β. MSU also elevated the mRNA level of GPX1 and SOD and L-NIL pretreatment suppressed MSU-induced the gene expression of these antioxidants enzymes (Fig. 3).
Mitogen-activated protein kinase (MAPK) expression
Phosphorylation of ERK1/2 and p38 was increased by MSU, which was reversed by L-NIL pretreatment. L-NIL alone did not alter the phosphorylation of ERK1/2 and p38 (Fig. 4). Phosphorylation of c-Jun-N-terminal kinase (JNK) was not affected by either MSU or L-NIL (data not shown).
Gene expression of iNOS and cytokines in cell lines
Gene expression of iNOS was increased in HDF, C2C12, and hFOB1.19 by MSU and L-NIL pretreatment attenuated iNOS mRNA expression in a dose dependent manner. Gene expression of iNOS was reduced by L-NIL treatment alone. MSU also increased mRNA expression of TNF-α and IL-1β in HDF, C2C12, and hFOB1.19, which were significantly attenuated by L-NIL in a dose dependent manner (Fig. 5).
DISCUSSION
The present study demonstrates that MSU induces edema and increases inflammatory cytokine expression in feet that is accompanied with increased iNOS expression. The selective iNOS inhibitor L-NIL suppresses the edema and cytokine expression. These results suggest that iNOS is involved in the development of gout.
The association between iNOS and rheumatoid arthritis or/and osteoarthritis has been extensively investigated and a large body of evidence suggests the important role of iNOS in these arthritis. The expression of iNOS is increased in the synovium, chondrocytes, vascular smooth muscle and inflammatory cells of rheumatoid arthritis and osteoarthritis subjects [20-24]. Moreover, suppression of iNOS expression in rheumatoid arthritis and osteoarthritis attenuates cell apoptosis, cytokine production and arthritis [20,25-27].
Despite extensive investigations as to the role of iNOS in rheumatoid arthritis and osteoarthritis, the relationship between iNOS and gouty arthritis has not been welldefined. Previous research has indicated that MSU increases iNOS expression in cells such as macrophages and chondrocytes in vitro [13,14,28]. Similar to this finding, we presently showed that MSU increased iNOS expression in fibroblast, myoblast, and osteoblast in vitro. Furthermore, we also observed that iNOS expression was increased in mice feet by MSU treatment, which is the first study showing increased iNOS expression by MSU treatment in vivo. The elevated levels of NO metabolites in plasma indirectly support increased iNOS expression by MSU. Additionally, the suppression of MSU-induced iNOS expression by L-NIL resulted in attenuated cytokine expression and edema. Consistent with our results, iNOS expression is enhanced in synovial tissues of gouty arthritis patients [13]. Together with this previous data, the current results support the notion that increased iNOS expression plays a causative role in the inflammation induced by MSU.
The mechanism by which MSU induces iNOS expression is unclear presently, but it is possible that MSU increases iNOS expression through MAPK pathways. MSU increases the MAPK subfamily member JNK, p38, and ERK1/2 [13,29,30], which are involved in a variety of MSU-induced pathological pathways [31]. ERK1/2 is involved in MSU-mediated transcriptional activation of IL-8 that functions as a neutrophil chemoattractant factor [29]. Inhibition of ERK1/2 or p38 reduces MSU-induced monocyte chemoattractant protein-1 in vascular smooth muscle cells [32]. MSU-induced iNOS expression is also mediated by p38 and ERK1/2 in chondrocytes and macrophages [13,14,28]. MSU activates p38 through the phosphorylation of proline-rich tyrosine kinase 2/focal adhesion kinase/protein paxillin, which increases iNOS expression and NO production [14]. Inhibition of ERK1/2 by specific inhibitor suppresses MSU-induced iNOS expression [13,28]. Consistent with these previous results, we presently observed that MSU increased the phosphorylation of ERK1/2 and p38. Additionally, the suppression of MSU-induced iNOS expression by L-NIL was accompanied with decreased the phosphorylation of both ERK1/2 and p38. Taken together, these results suggest the involvement of ERK/12 or/and p38 in MSU-induced iNOS expression. Although JNK is also increased in human monocyte cells line by MSU [33], JNK was not presently increased by MSU. Difference of species and experimental conditions could be the possible reasons.
We assume that increased iNOS expression could activate the production of inflammatory cytokines, which subsequently play a key role in gouty arthritis. Our notion is supported by the observation that suppression of MSU-induced iNOS expression by L-NIL presently attenuated cytokine expression and edema. A Previous study also demonstrated that the NO donor S-nitroso-acetyl penicillamine increases inflammatory cytokines such as TNF-α [34]. Gouty arthritis and administration of MSU crystal results in the production of a variety of inflammatory cytokines [35-37] and increased expressions of anti-inflammatory cytokines such as transforming growth factor β1, IL-1 receptor antagonist, IL-10, and soluble TNF receptor are correlated with spontaneous resolution of gouty arthritis [38].
Gouty arthritis is associated with oxidative stress [39] and the suppression of oxidative stress can reduce the symptoms [40]. Oxidative stress in gout may involve MSU-induced iNOS expression since elevated NO production from iNOS induces oxidative stress [12]. Presently, MSU increased the gene expression of anti-oxidant enzymes, which was normalized by L-NIL treatment. Antioxidant enzyme may be increased to protect the tissue from oxidative damage and attenuated oxidative stress by L-NIL normalizes the enzyme levels. Increased nitrotyrosine level by MSU was attenuated by L-NIL treatment presently, which also supports our theory.
In summary, MSU induces iNOS expression that is associated with increased expression of inflammatory cytokines, oxidative stress, and edema. ERK1/2 or/and p38 may mediates MSU-induced iNOS expression. Suppression of iNOS could be a new therapeutic target for gout.




 XML Download
XML Download