

 PDF
PDF ePub
ePub Citation
Citation Print
Print


INTRODUCTION
Strokes cause 9% of all deaths worldwide and are the third most common cause of death after ischemic heart disease and cancer. Moreover, given that 76% of people survive strokes in the United States and Europe, this condition is also the leading cause of adult disability [1]. The central goal of therapy in acute ischemic stroke is to preserve the area of oligemia in the ischemic penumbra. The area of oligemia can be preserved by limiting the severity of ischemic injury (i.e., neuronal protection) or by reducing the duration of ischemia (i.e., restoring blood flow to the compromised area). Despite a large number of studies on neuroprotection, no successful neuroprotective agents have shown to be clinically effective in the treatment of ischemic brain injury [2-4]. Therefore, the development of neuroprotective agents to prevent or treat this disorder is a current challenge.
Excitotoxicity, oxidative stress, and an excessive inflammatory response are all implicated in the pathogenesis of ischemic and reperfusion injury [5-7]. The major excitatory neurotransmitter glutamate plays important roles in the physiological functions of the central nervous system (CNS) and contributes to the pathogenesis of ischemia/hypoxia-induced neuronal injury [5,8]. Reactive oxygen species (ROS) have also been demonstrated to be overproduced in neural tissues during ischemia-reperfusion and have been indicated as one of the earliest and most important components of tissue injury in ischemic tissues [9]. The excessive production of ROS during cerebral ischemia-reperfusion can cause cellular damage via the oxidation of vital cellular components, such as lipids, proteins and DNA [10]. Other important targets of ROS are mitochondria, and mitochondrial dysfunction during ischemia/hypoxia results in neural injury [9,11]. Although the precise mechanisms underlying ischemia/hypoxia-induced neuronal cell death are not completely understood, there is a growing interest in establishing natural antioxidants and dietary supplements as potential therapeutic agents to combat ischemia-induced damage to the CNS [12-14].
Many studies have highlighted an important role for the neuroprotective action of dietary antioxidant components, including vitamins and phenolic compounds [15,16]. Anthocyanins are natural pigments belonging to the family of phenolic compounds and consist of a basic skeleton of either 2-phenylbenzopyrilium or flavylium glycoside. Anthocyanins are widely distributed in the human diet and are present in beans, vegetables and fruits. It has been reported that anthocyanin-rich berry extracts are protective in ischemia-reperfusion injury models of the heart [17], liver [18], brain [19], and in a traumatic spinal cord injury model [20]. Berry anthocyanins have also been shown to be protective against oxidative stress-induced PC12 cell death [19,21]. Furthermore, in folk medicine, mulberry (Morus alba L.) fruit has been used to treat and prevent diabetes, inflammation and fatigue. Therefore, mulberry anthocyanin may have protective effects against the ischemia-induced neuronal damage associated with excitotoxicity, oxidative stress, and inflammation. However, the neuroprotective activity of anthocyanin extracted from mulberry, known to contain only one type of anthocyanin, namely cyanidin-3-glucoside (C3G) [22], has not been examined in experimental models of oxygen-glucose deprivation (OGD) or in glutamate-induced excitotoxic cell death in primary cortical neurons. In this study, we, therefore, investigated the neuroprotective potential of C3G fraction extracted from mulberry fruit using a well-characterized culture of serum-free, highly pure rat primary cortical neurons.
Go to : 
METHODS
Materials
The tissue culture dishes and plates were purchased from TPP (Trasadingen, Switzerland). Dulbecco's modified Eagle medium (DMEM), Hank's balanced salt solution (HBSS), Neurobasal medium, supplement B27, supplement B27 without antioxidants, glutamine, penicillin/streptomycin, fetal bovine serum (FBS) and trypsin were all purchased from GIBCO BRL (Grand Island, NY). The dye rhodamine-123 was purchased from Molecular Probes (Eugene, OR), and L-glutamic acid was purchased from TOCRIS bioscience (Ellisville, MO). All other reagents were obtained from Sigma-Aldrich Co. (St. Louis, MO), unless indicated otherwise.
Extraction of C3G fraction from mulberry fruits
C3G fraction from mulberry fruit was prepared at Rural Development Administration, Suwon, Korea [23]. Briefly, following the addition of 0.1% citric acid-70% ethanol, fresh mulberry fruits were crushed manually and mixed three times at room temperature. The colored solution was immediately collected and filtered. The solution was subsequently evaporated using a large-scale evaporation system. C3G concentrate was then mixed with dextrin and freeze-dried. The lyophilized mulberry C3G fraction was stored at -70℃ until use.
Drug preparation and treatment
The stock solution of mulberry C3G fraction was prepared fresh prior to every experiment at a concentration of 50 mg/ml in 100% dimethyl sulfoxide (DMSO). The mulberry C3G fraction was further diluted in Neurobasal media or in HEPES-buffered balanced salt solution (BSS) containing 5.4 mM KCl, 125 mM NaCl, 20 mM HEPES, 1.8 mM CaCl2, and 0.01 mM glycine, pH 7.35. The final concentration of DMSO was <0.02%, which did not affect cell viability. The stock solutions of glutamate and MK801 were prepared in distilled water. The MK801 was used at a concentration of 1µM throughout the study. The cells were treated with mulberry C3G fraction throughout the entire experiment i.e., from 24 h prior to the OGD insult until 24 h following OGD (Fig. 2A), unless otherwise indicated.
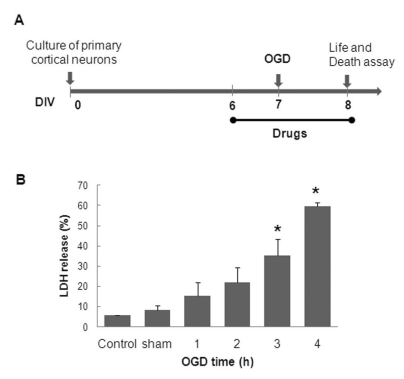 | Fig. 2The experimental design of the OGD-model in rat primary cortical neurons (A). OGD-duration-dependent neuronal cell death (B). DIV-7 rat primary cortical neurons were transiently placed in an anaerobic chamber for the indicated duration (1~4 h), and neuronal injury was assessed using LDH release assay 24 h following the simulated reperfusion. The values are given as the mean±S.E.M. of at least four independent experiments with different culture batches; *p<0.01 compared with the sham.
|
Primary culture of rat cortical neurons
Primary cortical neurons were prepared from Sprague-Dawley rats (Koatech, Gyeonggi, Korea) at 17 embryonic days according to a previously established method [24,25] with few modifications. Briefly, cerebral cortices from fetal rat brains (free of meninges, olfactory bulbs, striata and hippocampi) were pooled, chopped in Ca2+- and Mg2+-free HBSS and incubated with 0.025% trypsin for 10 min at 37℃. The enzymatic digestion was terminated by mixing the suspension with an equal volume of DMEM supplemented with 10% FBS. The cells were passed through cell strainers (BD Falcon, Bedford, MA) and collected by brief centrifugation. The dissociated cells were plated on 24-well plates (pre-coated with 10µg/ml poly-L-lysine) at a density of 3.2×105 cells/well in seeding medium consisting of Neurobasal medium supplemented with 2% B27, 0.5 mM glutamine, 25µM glutamate, 50 units/ml penicillin, and 50µg/ml streptomycin. The cultures were maintained at 37℃ in a humidified incubator with 5% CO2/95% air (normoxia). One day following plating, the seeding medium was removed and replaced with maintenance medium (seeding medium without glutamate) and refreshed twice a week. Neurons are the principal surviving cell type under these serum-free culture conditions [26]. These cultures were composed of more than 99% neurons, as determined by immunocytochemical staining with mouse anti-neuron specific nuclear protein (NeuN) and mouse anti-glial fibrillary acidic protein (GFAP) antibodies (Fig. 1). Each of the experiments was performed using Neurobasal medium (without phenol red) containing 2% antioxidant-free B27 supplement to rule out the anti-oxidant effects of the medium.
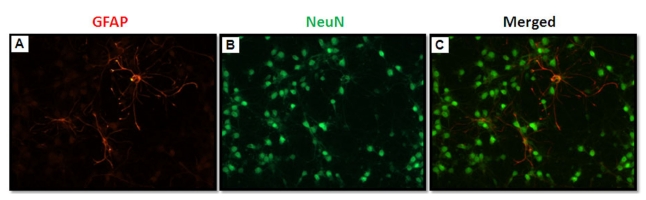 | Fig. 1Double fluorescent immunocytochemistry of DIV-7 rat primary cortical neurons in serum-free Neurobasal medium supplemented with B27. The cells were identified as astrocytes using an anti-GFAP antibody (red; A) or as neurons using an anti-NeuN antibody (green; B). Astrocyte contamination was <1% in this serum-free culture condition.
|
Oxygen-glucose deprivation
To simulate ischemic injury in vitro, primary cultured cortical neurons were subjected to OGD according to a previously established method [24,25] with few modifications. Briefly, at 7 days in vitro (DIV), the cells were washed twice with glucose-free BSS. To initiate OGD, deoxygenated glucose-free BSS (bubbled with 100% N2 for 20 min) was added to the cultures, and the plates were immediately transferred to an anaerobic chamber (Thermo Electron, Waltham, MA), which had been previously rinsed with an anaerobic gas mixture (5% CO2, 10% H2 and 85% N2). The cells were maintained in the chamber at 37℃ for 3.5 h to induce the ischemic insult. The oxygen concentration inside the anaerobic chamber was monitored occasionally with an anaerobic indicator strip (Oxoid Ltd., Hampshire, England). Following 3.5 h in anaerobic and glucose-free conditions, the OGD was terminated by changing the deoxygenated BSS to pre-warmed Neurobasal media containing antioxidant-free B27 supplements and returning the cells to a normal culture incubator. This reoxygenation incubation period is similar to the reperfusion period in in vivo ischemic injuries. Sham group neurons were washed twice with glucose-free BSS, incubated with BSS containing 25 mM glucose for 3.5 h in the normal culture incubator, and exposed to simulated reperfusion for 24 h.
Glutamate-induced neurotoxicity
At DIV-9, primary cortical neurons were washed with BSS containing 15 mM glucose. The cells were subsequently incubated with 50µM glutamate diluted in BSS for 10 min in the presence or absence of mulberry C3G fraction in a normal culture incubator. Following glutamate exposure, the neurons were washed twice with BSS and refreshed with pre-warmed Neurobasal media with or without mulberry C3G fraction for 24 h.
Assessment of neuronal injury
Neuronal injury was assessed using the lactate dehydrogenase (LDH) release and mitochondrial MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) reduction assays 24 h after OGD or glutamate exposure. Damage to the cell membrane of primary cortical neurons was quantitatively evaluated by determining the levels of LDH released from injured cells into the bathing medium 24 h following the end of OGD or glutamate exposure using the Cytotoxicity Detection Kit Plus (Roche, Mannheim, Germany) according to the manufacturer's instructions. Briefly, 100µl of sample medium was transferred from the culture wells to 96-well plates, mixed with 100µl of the reaction mixture provided by the kit, and incubated for 15 min at room temperature in the dark. After adding 50µl of stop solution, the absorbance (Abs) of the sample at 490 nm was determined using a multilabel counter system (Perkin Elmer, Boston, MA). The medium of completely lysed cells was defined as 100% LDH release. The absorbance values of normal background (normal media plus reaction mixer) were subtracted from values from the treatment and lysed groups. LDH (%) release was determined by the following ratio: Abs490 in the sample medium/Abs490 in the lysed cell medium. Using an MTT reduction assay, neuronal cell viability was estimated. This determination is based on the cleavage of yellow MTT salt into purple formazan by the mitochondrial dehydrogenase enzyme in viable cells. Briefly, following 24 h simulated reperfusion, fresh medium containing the MTT salt at a final concentration of 0.25 mg/ml was added to cultures and incubated for 1 h in a normal culture incubator. Following incubation, the medium was aspirated and DMSO was added to dissolve the insoluble purple formazan product into a colored solution, which was analyzed at a wavelength of 570 nm using a multilabel counter system (Perkin Elmer, Boston, MA). The absorbance of the formazan formed in the un-treated cells (controls) was defined as 100% viability.
Measurement of mitochondrial membrane potential (MMP)
The change in the MMP was measured in live primary cortical neurons using the fluorescent dye rhodamine-123. Primary cortical neurons grown on 24-well plates were loaded with 5µM rhodamine-123 in BSS. The cells were incubated for 10 min at 37℃ in a normal culture incubator and washed three times with BSS. The fluorescence signal of rhodamine-123 was read using a multilabel counter system (Perkin Elmer, Boston, MA). The background fluorescence signal of rhodamine-123 was also determined from wells without cells and was subtracted from the rhodamine-123 signals obtained from all sample wells. The MMP was calculated as the ratio of the fluorescent intensity of the sample cells to the intensity of the control (non-treated) cells. Fluorescent images of cells were captured at 20× using an Olympus IX70 inverted fluorescence microscope fitted with a digital color camera (Aomori Olympus, Tokyo, Japan).
Double immunofluorescence staining
To analyze the immunofluorescence data, primary cortical neurons grown on 12-mm glass coverslips (Deckglaser, Carolina Biologicals, Burlington, NC) were washed with Dulbecco's phosphate buffered saline (DPBS, pH 7.4), fixed with 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4) for 10 min, permeabilized with 0.1% Triton X-100 for 10 min, and washed with DPBS (5 min×3). After blocking with 10% normal goat serum for 1 h at room temperature, the cells were incubated with the primary antibody overnight at 4℃. The primary and secondary antibodies were diluted with 3% normal goat serum, and care was taken to avoid their exposure to light during the experiment. First, the cells were incubated with mouse anti-GFAP (1:500) (Chemicon, Temecula, CA) primary antibody overnight. The next day, the cells were washed and incubated with anti-mouse secondary antibody conjugated with Cy3 (1:500; Jackson ImmunoResearch, West Grove, PA) for 1 h. After washing, the cells were incubated with mouse anti-NeuN (1:200) antibody conjugated with Alexa-Fluor488 (Chemicon, Temecula, CA) for 2 h at room temperature. Following the final DPBS (5 min×3) wash, the glass cover-slips were mounted onto saline-coated slides with the fluorescent mounting media ProLong™ Gold (Invitrogen, Grand Island, NY). The cells were examined under a fluorescence microscope (AxioImager, Carl Zeiss, Jena, Germany).
Statistical analyses
Each group consisted of 3~4 culture wells per experiment and each set of experiments was performed at least three times with different culture batches. The data are expressed as the mean±the standard error of the mean (S.E.M). All statistical analyses were performed using SPSS 13.0 software (SPSS Inc., Chicago, IL). Statistical significance was assessed using the one-way analysis of variance for multiple group comparisons followed by Tukey's post hoc test. A p-value of less than 0.05 was considered to be statistically significant.
Go to :
RESULTS
Oxygen-glucose deprivation-induced neuronal cell death
In this study, a serum-free, highly pure rat primary cortical neuron culture (Fig. 1) was used to examine the neuroprotective activity of C3G fraction extracted from mulberry. To mimic cerebral ischemia in vitro, we used an OGD model of primary neurons cultured on 24-well plates. Preliminary experiments indicated that DIV-10 neurons appeared to be more resistant to OGD-reperfusion injury than DIV-7 neurons (data not shown). DIV-7 cells were, therefore, used for a time-course study of OGD-induced cell death. The resultant neuronal injury was quantified using the LDH release assay following 24 h of simulated reperfusion. It was observed that neuronal cell death depended on the duration of the OGD (Fig. 2B). The observed cell death in the sham group was 8.9±1.1%, whereas exposure to OGD for 1, 2, 3, and 4 h increased cell death to 16.7±3.5%, 22.7±4.3%, 37±5.2%, and 61.3±2.1%, respectively. Therefore, DIV-7 neurons and 3.5 h of OGD were chosen to simulate ischemia in vitro in the subsequent experiments.
Neuroprotective effect of mulberry C3G fraction against in vitro ischemia-induced cell death
We examined whether C3G fraction extracted from mulberry was able to protect primary cortical neurons against OGD-induced neuronal cell death. In the first series of experiments, the effect of C3G fraction alone on cell viability was determined using the MTT reduction assay. The treatment of primary cortical neurons with 1, 5, 10, 20, and 50µg/ml mulberry C3G fraction did not affect cell viability (data not shown). The exposure of primary cortical neurons to OGD for 3.5 h and reperfusion for 24 h resulted in a significant decrease in cell viability (37.6±5.3% in the OGD group compared to 95.1±1.6% in the sham group), as determined by the MTT reduction assay (Fig. 3A). On the other hand, treatment of neurons with mulberry C3G fraction at concentrations of 5 and 10µg/ml provided neuroprotection, increasing cell viability up to 61.5±8.0% and 61.4±7.3%, respectively. Moreover, to examine ischemia-induced membrane damage, we assessed the protective effect of mulberry C3G fraction on OGD-induced cellular damage using the LDH release assay. Treatment with mulberry C3G fraction at concentrations of 5 and 10µg/ml decreased LDH release to 30.8±3.1% and 29.9±2.6%, respectively, compared to 47.2±3.4% in the OGD-reperfusion group (Fig. 3B), thereby indicating that mulberry C3G fraction significantly attenuates neuronal membrane damage.
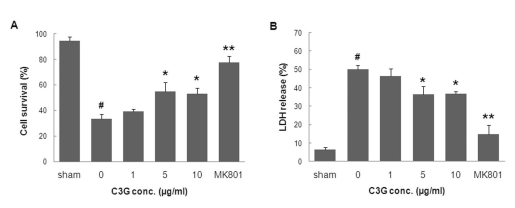 | Fig. 3The neuroprotective effect of mulberry C3G fraction against OGD-induced neuronal cell death. DIV-7 rat primary cortical neurons were exposed to OGD for 3.5 h with or without mulberry C3G fraction. Cell survival and death were measured 24 h following simulated reperfusion using the MTT reduction (A) and LDH release assays (B), respectively. The values are given as the mean±S.E.M. of at least six independent experiments. #p<0.01 compared with the sham. *p<0.05 and **p<0.01 compared with OGD without mulberry C3G fraction.
|
Glutamate induces excitotoxic cell death in primary cortical neurons
To examine the toxic effects of glutamate, primary cortical neurons cultured in vitro for different periods of time (DIV-7, -9 and -11) were exposed to various concentrations (25~500µM) of glutamate for 10 min. Twenty four hours following glutamate exposure, cell death was assessed using the LDH release assay. As shown in Fig. 4, glutamate-induced cell death was dependent on the period of time that the primary cortical neurons had been in culture. The LDH release assay indicated that glutamate-induced cell death in both DIV-9 and DIV-11 cells was greater than that observed in DIV-7 neurons. At a glutamate concentration of 50µM, cell death was induced at the rates of 23.9±2.2%, 41.3±5.0%, and 49.0±5.7% in DIV-7, -9 and -11 cultures, respectively. Because neuronal purity is compromised over 10 days in vitro, we chose DIV-9 cells to perform the subsequent glutamate toxicity experiments. At this stage, the culture is highly pure, and the cells are mature enough in terms of receptor expression and glutamate sensitivity [26,27]. The toxic effects of glutamate on DIV-9 and DIV-11 neurons did not linearly increase at concentrations higher than 50µM, and moderate cell deaths were observed at this concentration. Therefore, DIV-9 cultures and a glutamate concentration of 50µM were chosen as experimental conditions for the following studies of the neuroprotective effects of mulberry C3G fraction against excitotoxic cell death.
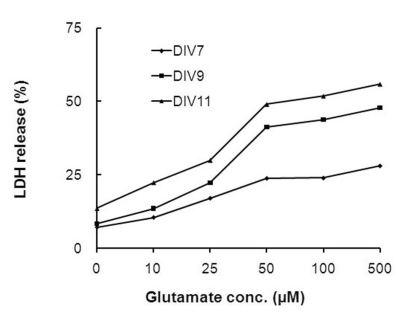 | Fig. 4The effects of different glutamate concentrations and culture durations on glutamate-induced neurotoxicity. Sister cultures of rat primary cortical neurons cultured for different periods of time were incubated with different concentrations of glutamate for 10 min. The resultant neuronal injury was assessed using the LDH release assay 24 h following the glutamate exposure. The values are given as the mean of at least four independent experiments with different culture batches.
|
Effect of mulberry C3G fraction on glutamate-induced primary cortical neuronal cell death
Having established that 50µM of glutamate induces neurotoxicity in cortical neurons, we examined whether mulberry anthocyanin has a protective effect against glutamate-induced toxicity in DIV-9 neurons. Primary cultured cortical neurons treated with or without various concentrations of mulberry C3G fraction (1~10µg/ml) and the non-competitive NMDA receptor antagonist MK801 (1µM) were exposed to 50µM of glutamate for 10 min. Following 24 h of glutamate exposure, neuronal injury was measured using the MTT reduction and LDH release assays. As illustrated in Fig. 5A, the treatment of primary cortical neurons with MK801 increased neuronal cell viability (90.3±3.1% vs. 53.4±1.78%) compared to glutamate-treated neurons in the absence of MK801. Treatment with MK801 also decreased glutamate-induced neuronal cell death, as determined by the observed reduction of LDH release (11.6±1.2% vs. 38.8± 4.6%) (Fig. 5B) compared to glutamate-treated neurons in the absence of MK801. Moreover, cell viability was 58.2± 1.3% and 57.0±1.6% in the mulberry C3G fraction-treated group at 5 and 10µg/ml, respectively. The LDH release assay indicated that cell death was 36.7±4.6% and 38.0±5.4% in the mulberry C3G fraction-treated group at 5 and 10µg/ml, respectively. Neither cell viability nor cell death in the C3G fraction- plus glutamate-treated groups were significantly different from the respective groups treated with glutamate only. These results suggest that C3G fraction extracted from mulberry is not effective against glutamate-induced excitotoxic cell death in rat primary cortical neurons.
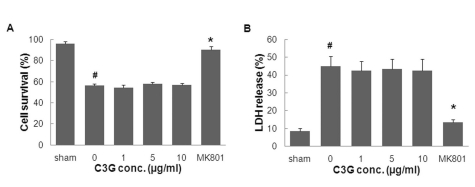 | Fig. 5The effects of mulberry C3G fraction on glutamate-induced neurotoxicity. DIV-9 cells were exposed to 50µM of glutamate with or without mulberry C3G fraction. Cell survival and death were measured 24 h following glutamate exposure using the MTT reduction (A) and LDH release assays (B), respectively. The values are given as the mean±S.E.M. of at least four independent experiments with different culture batches. #p<0.01 compared with the sham. *p<0.05 compared with glutamate without mulberry C3G fraction treatment.
|
Effect of mulberry C3G fraction on MMP
To examine the mechanism underlying the neuroprotective effects of C3G fraction extracted from mulberry, we investigated the effects of this extract on the MMP of primary cortical neurons exposed to OGD-reperfusion. The fluorescent dye rhodamine-123 was used to evaluate changes in MMP in primary cortical neurons, and the fluorescent intensity was expressed as the ratios of the fluorescence of treated neurons over that of control (un-treated) neurons. Relative to sham group (Fig. 6A), the fluorescence intensity in the OGD group was decreased (Fig. 6B). The treatment of neurons with 5µg/ml of mulberry C3G fraction preserved the signal intensity considerably, indicating a substantial ability of mulberry C3G fraction to maintain a polarized mitochondrial membrane potential (Fig. 6C) in primary cortical neurons subjected to OGD. Quantitative analyses indicated that the OGD insult resulted in a drastic decrease in the MMP signal intensity, which was 26.4±7.8% in the OGD-subjected group and 94.7±3.7% in the sham group. In contrast, treatment with mulberry C3G fraction resulted in a significant maintenance of the MMP signal intensity in primary cortical neurons exposed to OGD-reperfusion. Signal intensity values were 59.6±8.2% and 59.7±7.8% at C3G concentrations of 5 and 10µg/ml, respectively (Fig. 6D).
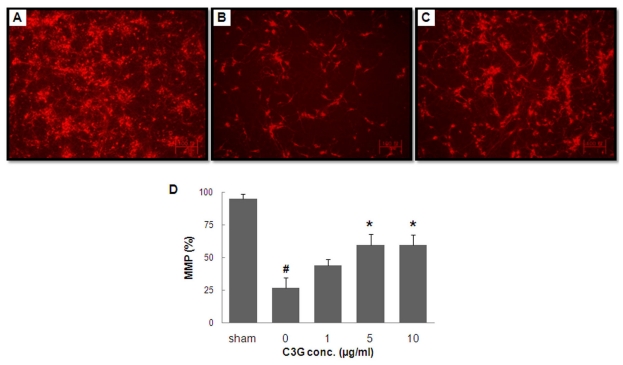 | Fig. 6The effects of mulberry C3G fraction on MMP maintenance in primary cortical neurons subjected to OGD. DIV-7 cells were exposed to OGD for 3.5 h with or without mulberry C3G fraction treatment. Following 24 h of simulated reperfusion, the MMP was measured using the fluorescent dye rhodamine-123. Representative fluorescent images of sham-treated cells (A), cells subjected to OGD only (B) and cells subjected to OGD with 5µg/ml of mulberry C3G fraction (C). The scale bar is 100 mm. The bar graph (D) summarizes the fluorescence measurements from different groups. The values are given as the mean±S.E.M. of at least three independent experiments with different culture batches. #p<0.01 compared with the sham. *p<0.05 compared with OGD without mulberry C3G fraction treatment.
|
Go to :
DISCUSSION
The cellular and molecular cascades underlying ischemic injury are multifaceted and complex [7]. Excitotoxicity, oxidative stress, and an excessive inflammatory response have been implicated in the progressive neuronal injury and cell death observed post-ischemia [9,11]. ROS overproduction has been identified as one of the earliest and most important mediators of tissue injury during the ischemia-reperfusion insult. For this reason, and because of their positive roles against ischemia-reperfusion injury, much attention has recently been focused on natural antioxidants. Anthocyanin-rich berry extracts and their major component C3G are known to possess antioxidant and anti-inflammatory activity in various disease models [28-31]. In this study, we evaluated the neuroprotective activity of C3G fraction extracted from mulberry in models of OGD-reperfusion and glutamate-induced cell death in rat primary cortical neurons. The results of this study indicate that mulberry C3G fraction is neuroprotective against OGD-induced primary cortical neuronal death. Furthermore, this protection was conferred, at least in part, via the inhibition of membrane damage and the preservation of the MMP and mitochondrial function.
The OGD model is considered to be the closest simulation of cerebral ischemia for studying ischemia in vitro and has been used in numerous ischemia-related studies [32-34]. In this study, highly pure cultures of primary cortical neurons were prepared from embryonic day 17 fetal rat brains. The neurons were plated onto pre-coated culture surfaces with Neurobasal media (favorable to neurons) without serum supplements. Suitable conditions were subsequently established to induce progressive neuronal death following in vitro ischemia. It was found that some neurons exhibited a swollen morphology and branched off immediately following OGD. Widespread neuronal degeneration occurred over the reperfusion period, despite the resupply of normal oxygen and glucose levels. In this study, we observed that the treatment of primary cortical neurons with mulberry C3G fraction prevented OGD/reperfusion-induced neuronal degeneration. It has been reported that the presence of glial cells in the culture and/or serum supplementation in the media regulates and promote cell survival in primary neuron cultures [35-38]. Because our culture was highly pure neuronal and was serum-free, it can be inferred that mulberry C3G fraction afforded the neuroprotection observed in primary cortical neurons against the damage induced by the OGD-reperfusion.
The events mediating cell death in the context of OGD include ATP depletion, excitotoxicity, and oxidative stress, all of which are associated with mitochondrial function [11]. Both oxidative stress and ATP depletion are reported to be involved in mitochondrial dysfunction, which is marked by the collapse of the mitochondrial trans-membrane potential. Furthermore, the loss of the MMP alters mitochondrial membrane permeability. We examined MMP changes in primary cortical neurons that were subjected to the OGD-reperfusion using rhodamine-123, a cell permeable cationic fluorescent dye that can be used to directly measure the MMP. Our results indicate that mulberry C3G fraction prevents OGD-reperfusion-induced MMP loss in primary cortical neurons. These results suggest that the protection of mitochondria from OGD-induced stress is a possible mechanism by which mulberry C3G fraction blocks cell death. In this regard, Kang et al. [19] reported that C3G, the major anthocyanin component in mulberry fruit, exhibited a significant neuroprotective effect against OGD-induced PC12 cell death. In support of our findings, this group also found that crude mulberry extract and C3G were neuroprotective against in vivo cerebral ischemia in mouse and H2O2-induced oxidative stress in PC12 cells. Surprisingly, this group did not report a protective effect of crude extract of mulberry against OGD-induced PC12 cell death, even at a conconcentration of 10µg/ml. These conflicting findings may be due to the different cell types, extraction solvents, protocols, and/or drug treatment schedules used. The prior researchers used immortal PC12 cells, whereas we used serum-free primary cortical neurons, which are widely used to mimic cerebral ischemia in vitro. Another crucial difference between these two studies was the use of solvents to extract anthocyanin from the mulberry fruit. In their study, methanol was used for anthocyanin extraction. Although methanol is more efficient to extraction of anthocyanins [23], methanolic extract is not considered edible. However, in our study, citric acid-ethanol was used to extract C3G from mulberry fruit. This substance is edible and has the potential to be used as a supplement following further studies in animal models and/or clinical trials. Moreover, we observed a neuroprotective effect of mulberry C3G fraction at a concentration of 5µg/ml in more severe OGD settings (3.5 h vs. 2 h, and ≈37% vs. ≈55% of cell viability), which may be due to the differences in drug-treatment schedules. Kang et al. treated PC12 cells with mulberry crude extract and C3G during only 2 h of OGD, whereas we treated primary neurons with C3G fraction beginning 24 h prior to the OGD insult until 24 h following the OGD. Therefore, it appears that pre- and post-treatments of neurons with mulberry C3G fraction may contribute to the neuroprotective effect of this extract at a relatively lower concentration. However, the additive neuroprotective effect of other component(s) present in crude C3G fraction cannot be excluded, given that the mulberry C3G fraction used in our study was not 100% pure.
Glutamate-induced excitotoxicity can also contribute to neuronal damage and degeneration following in vivo cerebral ischemia [39,40] and in vitro hypoxia/ischemia models, including OGD [8,34,41]. To determine the mechanism of the observed neuroprotective effect induced by mulberry C3G fraction against OGD, we, therefore, tested the effect of mulberry C3G fraction on glutamate-induced primary neuronal cell death. It was found that mulberry C3G fraction did not inhibit primary cortical neuronal death following glutamate treatment. To our knowledge, there have been no reports demonstrating the effects of anthocyanins, including C3G, against glutamate-induced excitotoxicity in vitro. However, one in vivo study reported that bilberry extracts inhibited NMDA-induced retinal damage in mice [30]. Because bilberry extract consists of approximately 15 different anthocyanins [30,42], it is possible that anthocyanin components other than C3G may exhibit anti-excitotoxic activity. However, further studies are required to verify the effects of anthocyanins, including the actions of C3G against excitotoxic insults.
Many studies have demonstrated the beneficial effects of anthocyanins, including C3G, against numerous degenerative diseases [17-21,28-31]. To our knowledge, however, no reports have demonstrated adverse effects of C3G. However, one study by Ziberna et al. (2010) reported cardiotoxic effects of relatively high concentrations of anthocyanins extracted from bilberry fruit [43]. It was shown that at concentrations of 0.1~5µg/ml, anthocyanins extracted from bilberries conferred cardioprotective effects against ischemia-reperfusion injury in isolated rat hearts. However, concentrations of 20~50µg/ml significantly increased LDH release in ischemic hearts compared to control, untreated ischemic hearts. In our study, concentrations of 5~10µg/ml mulberry C3G fraction exhibited neuroprotective activity against OGD-induced death in primary neurons. Because, we did not examine the neuroprotective activity of mulberry C3G fraction at concentrations greater than 10µg/ml, it is possible that higher concentrations may result in diminished neuroprotection or even facilitate toxicity in the context of ischemia-reperfusion. In fact, our preliminary cytotoxicity test indicated that the incubation of rat primary cultured cortical neurons with much higher concentrations (100µg/ml) of mulberry C3G fraction results in neuronal death, even under physiological conditions (data not shown). Therefore, further studies are required to determine whether there are any adverse effects linked to higher concentrations of mulberry C3G fraction in the context of ischemia-reperfusion.
In conclusion, our study demonstrates the protective effects of C3G fraction extracted from mulberry fruits against OGD-reperfusion-induced neuronal injury. Our results also indicate that these neuroprotective effects are partly mediated by the maintenance of mitochondrial function and MMP but not by the inhibition of glutamate-induced excitotoxicity in rat primary cortical neurons. Although further studies of the effects of anthocyanins are required in different models of hypoxia/ischemia and other neurodegenerative diseases, the present study provides important information regarding the neuroprotective activities of mulberry C3G fraction against ischemia-induced neuronal cell death.
Go to :
 XML Download
XML Download