

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
A non-steroidal anti-inflammatory drug (NSAID) has many adverse effects including cardiovascular (CV) risk. Diclofenac among the nonselective NSAIDs has the highest CV risk such as congestive heart failure, which resulted commonly from the impaired cardiac pumping due to a disrupted excitation-contraction (E-C) coupling. We investigated the effects of diclofenac on the L-type calcium channels which are essential to the E-C coupling at the level of single ventricular myocytes isolated from neonatal rat heart, using the whole-cell voltage-clamp technique. Only diclofenac of three NSAIDs, including naproxen and ibuprofen, significantly reduced inward whole cell currents. At concentrations higher than 3 μM, diclofenac inhibited reversibly the Na+ current and did irreversibly the L-type Ca2+ channels-mediated inward current (IC50=12.89±0.43 μM) in a dose-dependent manner. However, nifedipine, a well-known L-type channel blocker, effectively inhibited the L-type Ca2+ currents but not the Na+ current. Our finding may explain that diclofenac causes the CV risk by the inhibition of L-type Ca2+ channel, leading to the impairment of E-C coupling in cardiac myocytes.
Go to : 
References
Alves D., Duarte I. Involvement of ATP-sensitive K+ channels in the peripheral antinociceptive effect induced by dipyrone. Eur J Pharmacol. 444:47–52. 2002.
Asomoza-Espinosa R., Alonso-Lopez R. Mixcoatl-Zecuatl T., Aguirre-Banuelos P., Torres-Lopez JE., Granados-Soto V. Sildenafil increases diclofenac antinociception in the formalin test. Eur J Pharmacol. 418:195–200. 2001.

Bodi I., Mikala G., Koch SE., Akhter SA., Schwartz A. The L-type calcium channel in the heart: the beat goes on. J Clin Invest. 115:3306–3317. 2005.
Bort R., Ponsoda X., Jover R., Gomez-Lechon MJ., Castell JV. Diclofenac toxicity to hepatocytes: a role for drug metabolism in cell toxicity. J Pharmacol Exp Ther. 288:65–72. 1998.
Brater DC. Renal effects of cyclooxygyenase-2-selective inhibitors. J Pain Sympt Management. 23:S15–S20. 2002.
Cha TJ., Ehrlich JR., Zhang L., Shi YF., Tardif JC., Leung TK., Nattel S. Dissociation between remodeling and ability to sustain atrial fibrillation during recovery from experimental congestive heart failure. Circulation. 109:412–418. 2004.
Dalla Libera L., Vescovo G., Volterrani M. Physiological basis for contractile dysfunction in the heart failure. Curr Pharm Design. 14:2572–2581. 2008.
Doering CJ., Zamponi GW. Molecular pharmacology of high voltage-activated calcium channels. J Bioenerg and Biomem. 35:491–505. 2003.
Fei XW., Liu LY., Xu JG., Zhang ZH., Mei YA. The non-steroidal anti-inflammatory drug, diclofenac, inhibits Na+ current in rat myoblasts. Biochem Biophys Res Commun. 346:1275–1283. 2006.
Ferrier GR., Howlett SE. Contractions in guinea-pig ventricular myocytes triggered by a calcium-release mechanism separate from Na+ and L-currents. J Physiol. 484:107–122. 1995.
Ferrier GR., Redondo IM., Mason CA., Mapplebeck C., Howlett SE. Regulation of contraction and relaxation by membrane potential in cardiac ventricular myocytes. Am J Physiol. 278:H1618–H1626. 2000.
Fu J., Gao J., Pi R., Liu P. An optimized protocol for culture of cardiomyocytes from neonatal rat. Cytotechnol. 49:109–116. 2005.
Graham DJ. COX-2 inhibitors, other NSAIDs, and cardiovascular risk: the seduction of common sense. J Am Med Assoc. 296:1653–1656. 2006.
Hobai IA., Howarth FC., Pabbathi VK., Dalton GR., Hancox JC., Zhu JQ., Howlett SE., Ferrier GR., Levi AJ. Voltage-activated Ca2+ release in rabbit, rat and guinea-pig cardiac myocytes, and modulation by internal cAMP. Pflugers Arch. 435:164–173. 1997.
Hombach V. Electrocardiography of the failing heart. Cardiol Clin. 24:413–426. 2006.
Howlett SE., Zhu JQ., Ferrier GR. Contribution of a voltage-sensitive calcium release mechanism to contraction in cardiac ventricular myocytes. Am J Physiol. 274:H155–H170. 1998.
Hudson M., Rahme E., Richard H., Pilote L. Risk of congestive heart failure with nonsteroidal anti-inflammatory drugs and selective cyclooxygenase 2 inhibitors: a class effect? Arthritis and Rheumatism. 57:516–523. 2007.
Katsube Y., Yokoshiki H., Nguyen L., Yamamoto M., Sperelakis N. L-type Ca2+ currents in ventricular myocytes from neonatal and adult rats. Can J Physiol Pharmacol. 76:873–881. 1998.
Kearney PM., Baigent C., Godwin J., Halls H., Emberson JR., Patrono C. Do selective cyclo-oxygenase-2 inhibitors and traditional non-steroidal anti-inflammatory drugs increase the risk of atherothrombosis? Meta-analysis of randomised trials. Br Med J. 332:1302–1308. 2006.
Larsen JK., Mitchell JW., Best PM. Quantitative analysis of the expression and distribution of calcium channel α1 subunit mRNA in the atria and ventricles of the rat heart. J Mol Cell Cardiol. 34:519–532. 2002.
Lee HM., Kim HI., Shin YK., Lee CS., Park M., Song JH. Diclofenac inhibition of sodium currents in rat dorsal root ganglion neurons. Brain Research. 992:120–127. 2003.
Lee JH., Gomora JC., Cribbs LL., Perez-Reyes E. Nickel block of three cloned T-type calcium channels: low concentrations selectively block a1H. Biophys J. 77:3034–3042. 1999.
Leucuta A., Vlase L., Farcau D., Nanulescu M. No effect of short term ranitidine intake on diclofenac pharmakinetics. Rom J Gastroenterol. 13:306–308. 2004.
Lindner M., Erdmann E., Beuckelmann DJ. Calcium content of the sarcoplasmic reticulum in isolated ventricular myocytes from patients with terminal heart failure. J Mol Cell Cardiol. 30:743–749. 1998.
Liu LY., Fei XW., Li ZM., Zhang ZH., Mei YA. Diclofenac, a nonsteroidal anti-inflammatory drug, activates the transient outward K+ current in rat cerebellar granule cells. Neuropharmacol. 48:918–926. 2005.
Maltsev VA., Sabbab HN., Undrovinas AI. Down-regulation of sodium current in chronic heart failure: effect of long-term therapy with carvediol. Cell Mol Life Sci. 59:1561–1568. 2002.
McGettigan P., Henry D. Cardiovascular risk and inhibition of cyclooxygenase: a systemic review of the observational studies of selective and nonselective inhibitors of cyclooxygenase-2. J Am Med Assoc. 296:1633–1644. 2006.
Morales MA., Inostroza L., Salazar T., Paeile C. Effects of clonixin on the electrical activity of cardiac pacemaker cells. Gen Pharmacol. 23:515–521. 1992.
Morales MA., Salazar T., Paeile C. Effects of flunixin and mefenamic acid on cardiac pacemaker cells. Structure-activity relationship and comparison with clonixin. Gen Pharmacol. 24:775–780. 1993.
Nawrath H., Klein G., Rupp J., Wegener JRW., Shainberg A. Open state block by fendiline of L-type Ca2+ channels in ventricular myocytes from rat heart. J Pharmacol Exp Ther. 285:546–552. 1998.
Ortiz MI., Torres-Lopez JE., Castaneda-Hernandez G., Rosas R., Vidal-Cantu GC., Granados-Soto V. Pharmacological evidence for the activation of K+ channels by diclofenac. Eur J Pharmacol. 438:85–91. 2002.
Perez-Reyes E. Molecular characterization of a novel family of low voltage-activated, T-type, calcium channels. J Bioenerg Biomem. 30:313–318. 1998.
Perez-Reyes E., Lee JH., Cribbs LL. Molecular characterization of two members of the T-type calcium channel family. Ann N Y Acad Sci. 868:131–143. 1999.
Pieske B., Maier LS., Bers DM., Hasenfuss G. Ca2+ handling and sarcoplasmic reticulum Ca2+ content in isolated failing and nonfailing human myocardium. Circ Res. 85:38–46. 1999.
Pinto JM., Boyden PA. Electrical remodeling in ischemia and infarction. Cardiovasc Res. 42:284–297. 1999.
Tan HL., Bink-Boelkens MT., Bezzina CR., Viswanathan PC., Beaufort-Krol GC., van Tintelen PJ., van den Berg MP., Wilde AA., Balser JR. A sodium channel mutation causes isolated cardiac conduction disease. Nature. 409:1043–1047. 2001.
Tonussi CR., Ferreira SH. Mechanism of diclofenac analgesia: direct blockade of inflammatory sensitization. Eur J Pharmacol. 251:173–179. 1994.
Waksman JC., Brody A., Phillips SD. Nonselective nonsteroidal anti-inflammatory drugs and cardiovascular risk: are they safe? Ann Pharmacother. 41:1163–1173. 2007.
Willis JV., Kendall MJ., Flinn RM., Thornhill DP., Welling PG. The pharmacokinetics of diclofenac sodium following intravenous and oral administration. Eur J Clin Pharmacol. 16:405–410. 1979.
Yang YC., Kuo CC. An inactivation stabilizer of the Na+ channel acts as an opportunistic pore blocker modulated by external Na+. J Gen Physiol. 125:465–481. 2005.
Zhu JQ., Ferrier GR. Regulation of a voltage-sensitive release mechanism by Ca2+-calmodulin dependent kinase in cardiac myocytes. Am J Physiol. 279:H2104–H2115. 2000.
Go to :
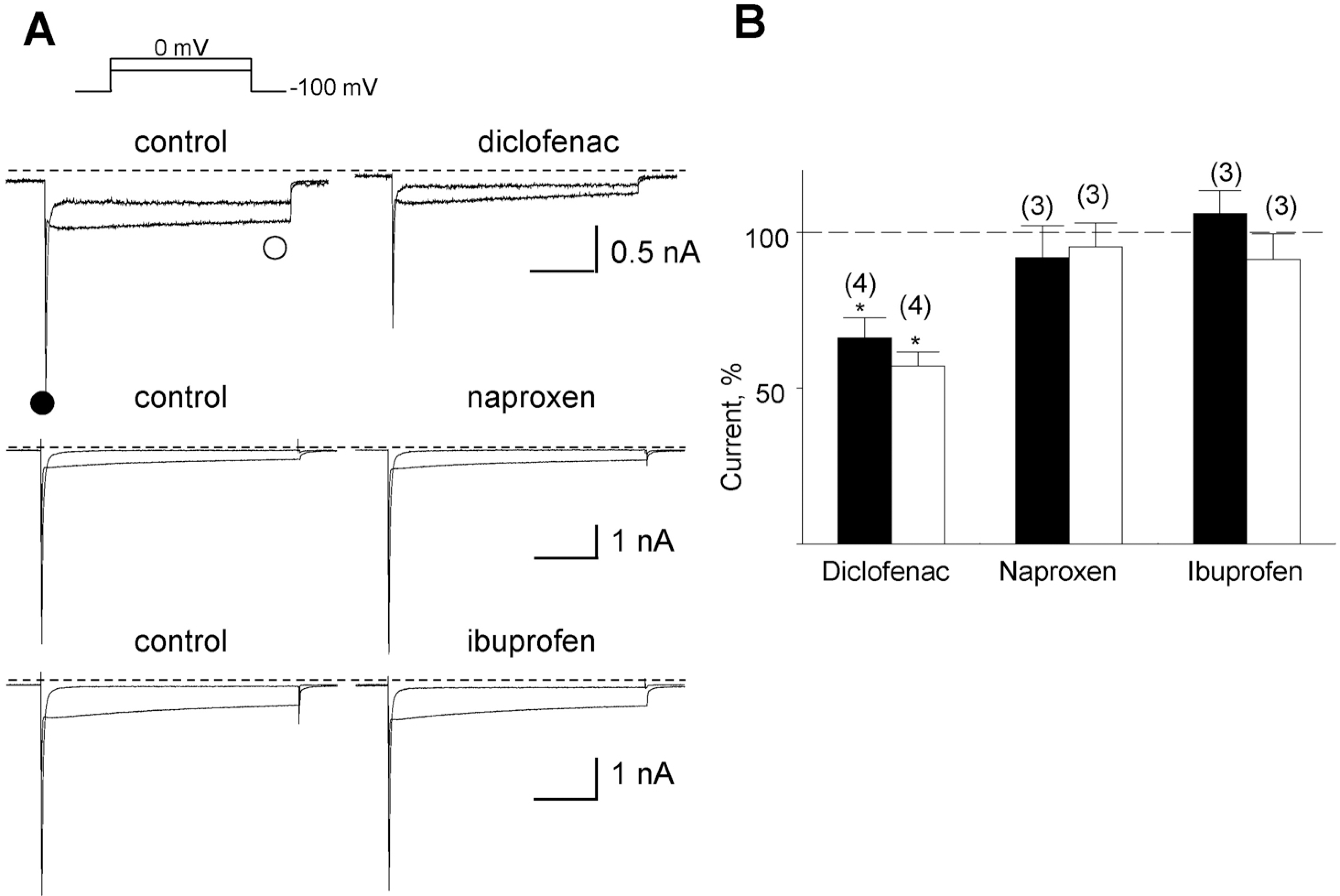 | Fig. 1.Inhibition of the Na+- and the Ca2+-sensitive inward current by three NSAIDs. (A) Representative currents before and after application of the drugs denoted above the corresponding trace. The drugs were applied at a concentration of 10 μM each. The amplitudes of the initial transient component and the slowly decayed components were measured at positions marked by closed () and open circles (❍), respectively. (B) Summary of the normalized data for the effect of drugs on two components. Relative inhibition (%) for the Na+-sensitive initial transient (black bar) and the nifedipine-sensitive slowly decayed components (open bar) are shown with number of observations. Data were normalized to currents measured before application of each drug. |
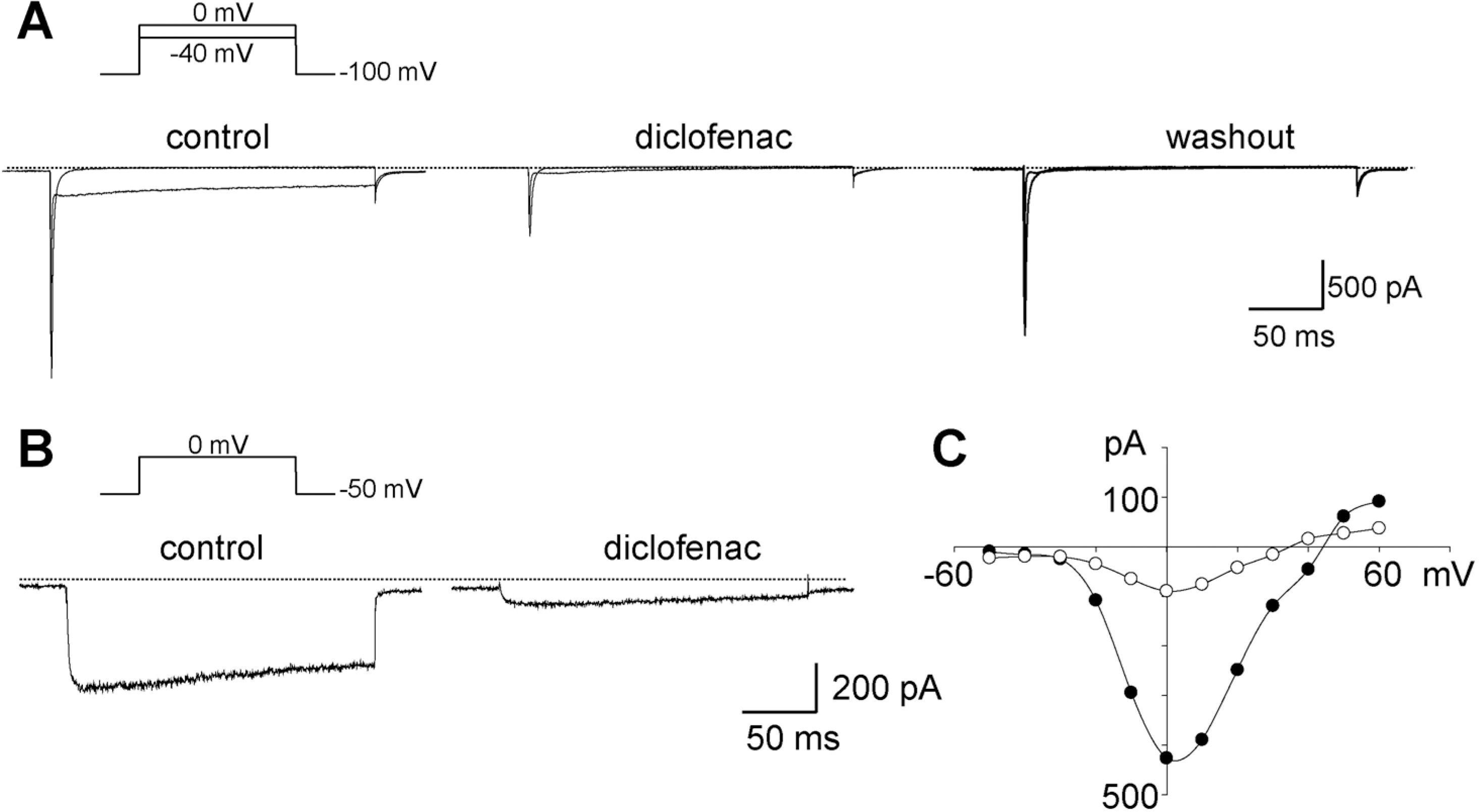 | Fig. 2.Representative traces of whole-cell currents elicited by step depolarizations in single cardiac myocytes. (A) Inhibition of the inward current induced by diclofenac. Changes in whole-cell currents evoked at –40 and 0 mV from a holding potential of –100 mV in bath solution containing 140 mM Na+ before (left) and after adding diclofenac (middle), and following washout (right), respectively. Dotted lines in A and B indicate the zero current level. (B) Currents induced by depolarization as indicated above the traces, in Na+-free bath solution before and after the addition of diclofenac. (C) Current-voltage relationship measured from the peak current of the traces in panel B. Diclofenac of 100 μM was applied. Outward components were not detected due to the presence of Ba2+, instead of Ca2+ in the bath. |
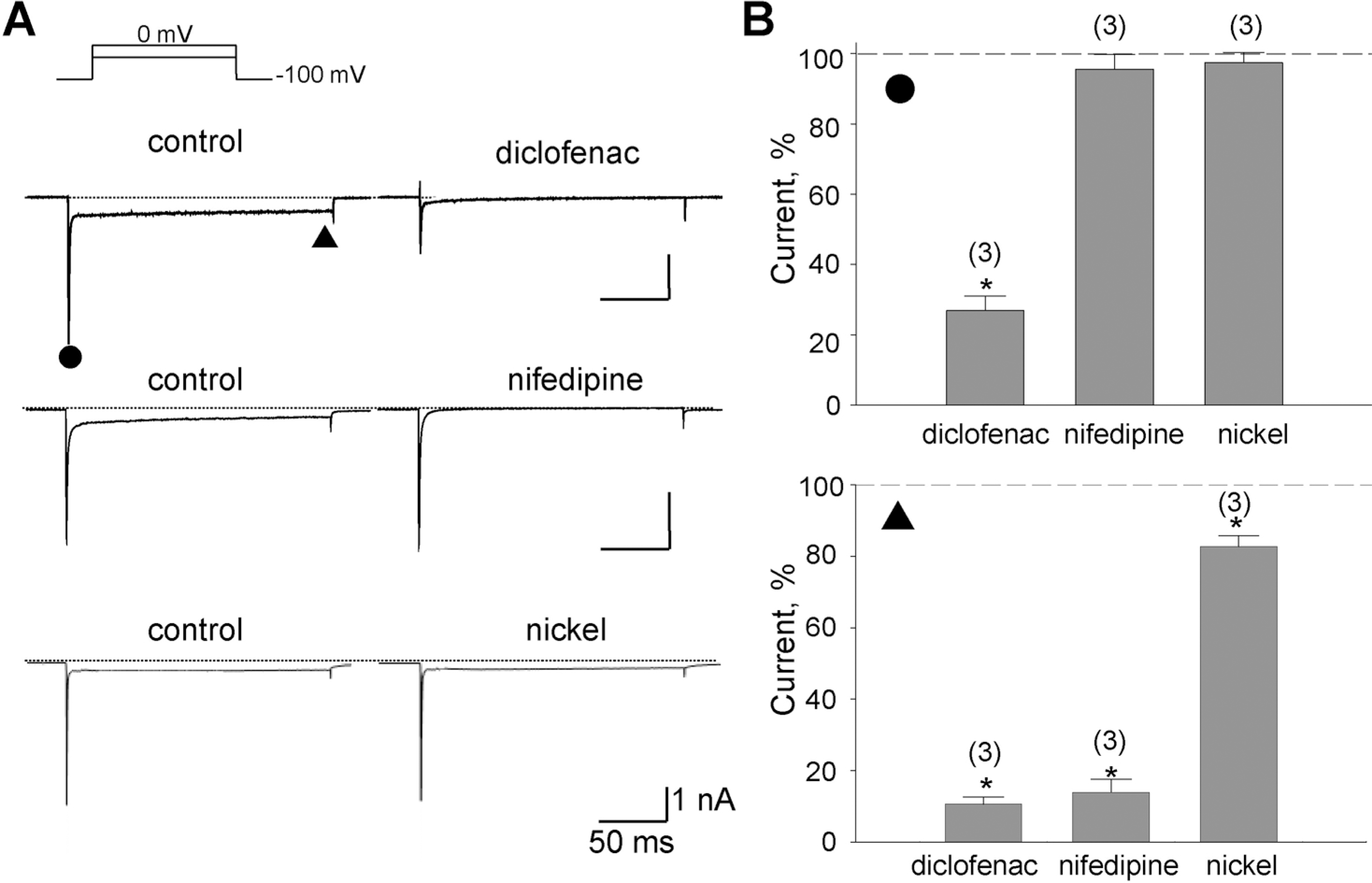 | Fig. 3.Inhibition of the Na+ and the Ba2+ components by diclofenac. (A) Representative currents inhibited by drugs denoted above the right trace. With the application of diclofenac (100 μM), nifedipine (1 μM), or nickel (300 μM), reduced currents were shown on the right. The amplitudes of the initial transient component and the slowly decayed components were measured at the positions marked by the closed circle () and triangle (▴), respectively. (B) Summary of the normalized data for the effect of drugs on the two components. Relative inhibitions (%) of the Na+-sensitive initial transient and the nifedipine-sensitive components are shown in upper and lower panel with number of observations, respectively. Data were normalized to currents measured before application of each drug. Scale bars are equal to 1 nA and 50 ms. |
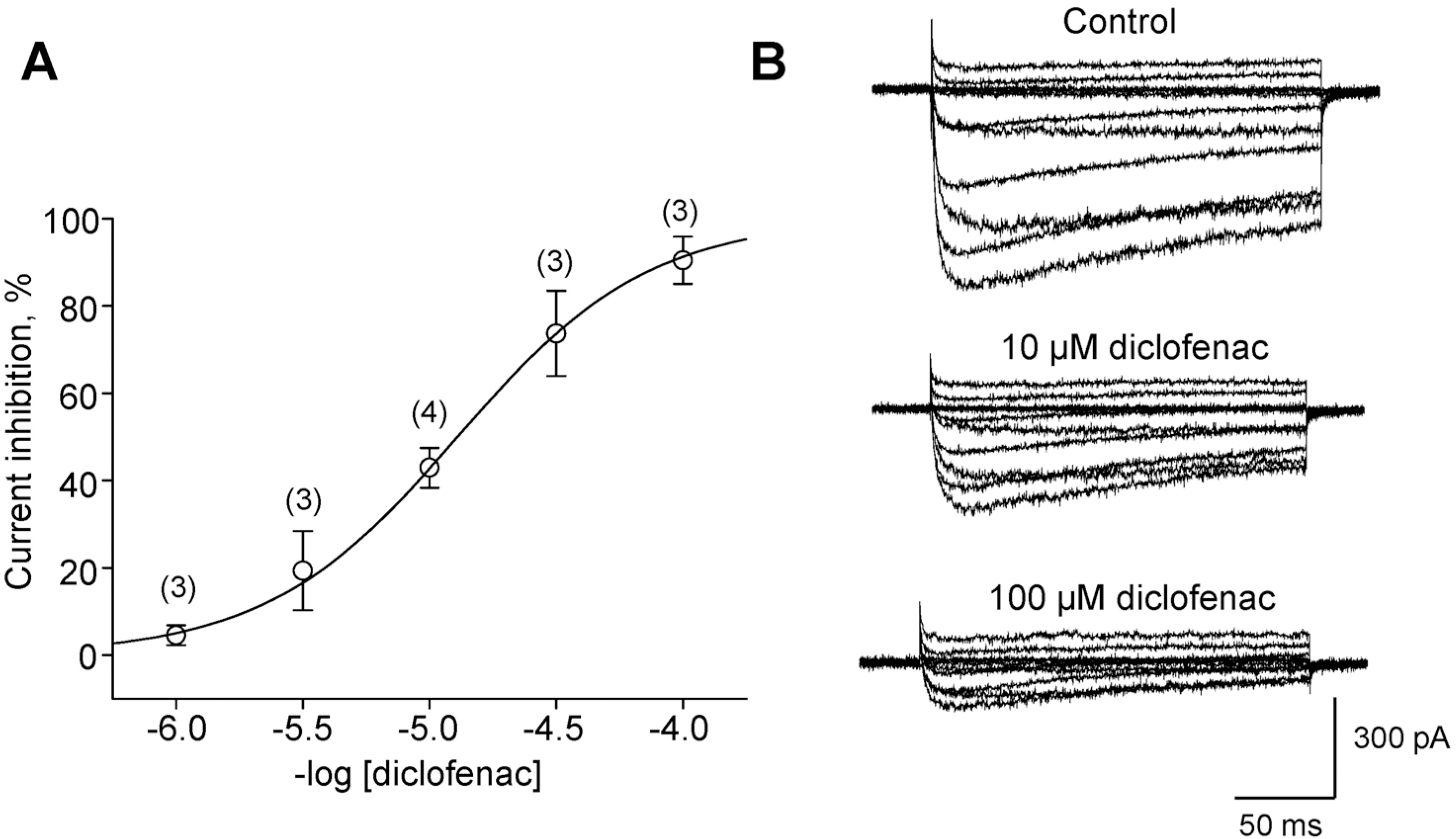 | Fig. 4.Dose-dependent inhibition of the L-type current by diclofenac. (A) Dose-response relationship of the inhibitory effect of diclofenac on peak L-type (IBa) currents in cardiomyocytes, with the numbers of cells. The molar concentration of diclofenac is given. (B) Representative traces of L-type currents reduced by diclofenac. Step depolarizations were applied from HP of –50 mV to +60 mV in 10 mV increments. |
 XML Download
XML Download