

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Abstract
Heat shock proteins (HSPs) serve as molecular chaperones and play a role in cell protection from damage in response to stress stimuli. The aim of this article is to investigate whether HSP22 affects IL-8 expression in vascular smooth muscle cells (VSMCs), and which cellular factors are involved in the HSP-mediated IL-8 induction in that cell type in terms of mitogen activated protein kinase (MAPK) and transcription element. Exposure of aortic smooth muscle cells (AoSMCs) to HSP22 not only enhanced IL-8 release but also induced IL-8 transcript via promoter activation. HSP22 activated ERK and p38 MAPK in AoSMCs. HSP22-induced IL-8 release was inhibited by U0126, but not by SB202190. A mutation in the IL-8 promoter region at the binding site of NF-κB, but not AP-1 or C/EBP, impaired promoter activation in response to HSP22. Delivery of IκB, but not dominant negative c-Jun, lowered HSP22-induced IL-8 release from AoSMCs. These results suggest that HSP22 induces IL-8 in VSMCs via ERK1/2, and that transcription factor NF-kB may be required for the HSP22-induced IL-8 up-regulation.
Go to : 
REFERENCES
Aiyar N., Disa J., Ao Z., Ju H., Nerurkar S., Willette RN., Macphee CH., Johns DG., Douglas SA. Lysophosphatidylcholine induces inflammatory activation of human coronary artery smooth muscle cells. Mol Cell Biochem. 295:113–120. 2007.

Akira S., Takeda K. Toll-like receptor signalling. Nat Rev Immunol. 4:499–511. 2004.
Boisvert WA., Santiago R., Curtiss LK., Terkeltaub RA. A leukocyte homologue of the IL-8 receptor CXCR-2 mediates the accumulation of macrophages in atherosclerotic lesions of LDL receptor-deficient mice. J Clin Invest. 101:353–363. 1998.
Brown PH., Chen TK., Birrer MJ. Mechanism of action of a dominant-negative mutant of c-Jun. Oncogene. 9:791–799. 1994.
de Jong WW., Leunissen JA., Voorter CE. Evolution of the alpha-crystallin/small heat-shock protein family. Mol Biol Evol. 10:103–126. 1993.
Gerszten RE., Garcia-Zepeda EA., Lim YC., Yoshida M., Ding HA., Gimbrone MA., Jr. Luster A D., Luscinskas FW. Rosenzweig A. MCP-1 and IL-8 trigger firm adhesion of monocytes to vascular endothelium under flow conditions. Nature. 398:718–723. 1999.
Ho FM., Kang HC., Lee ST., Chao Y., Chen YC., Huang LJ., Lin WW. The anti-inflammatory actions of LCY-2-CHO, a carbazole analogue, in vascular smooth muscle cells. Biochem Pharmacol. 74:298–308. 2007.
Huo Y., Weber C., Forlow SB., Sperandio M., Thatte J., Mack M., Jung S., Littman DR., Ley K. The chemokine KC, but not monocyte chemoattractant protein-1, triggers monocyte arrest on early atherosclerotic endothelium. J Clin Invest. 108:1307–1314. 2001.
Inoue T., Komoda H., Nonaka M., Kameda M., Uchida T., Node K. Interleukin-8 as an independent predictor of long-term clinical outcome in patients with coronary artery disease. Int J Cardiol. 124:319–325. 2008.
Jung YD., Fan F., McConkey DJ., Jean ME., Liu W., Reinmuth N., Stoeltzing O., Ahmad SA., Parikh AA., Mukaida N. Role of P38 MAPK, AP-1, and NF-kappaB in interleukin-1beta-induced IL-8 expression in human vascular smooth muscle cells. Cytokine. 18:206–213. 2002.
Kawai T., Akira S. Toll-like receptor downstream signaling. Arthritis Res Ther. 7:12–19. 2005.
MacRae TH. Structure and function of small heat shock/alpha-crystallin proteins: established concepts and emerging ideas. Cell Mol Life Sci. 57:899–913. 2000.
Moreau M., Brocheriou I., Petit L., Ninio E., Chapman MJ., Rouis M. Interleukin-8 mediates down-regulation of tissue inhibitor of metalloproteinase-1 expression in cholesterol-loaded human macrophages: relevance to stability of atherosclerotic plaque. Circulation. 99:420–426. 1999.
Perschinka H., Mayr M., Millonig G., Mayerl C., van der Zee R., Morrison SG., Morrison RP., Xu Q., Wick G. Cross-reactive B-cell epitopes of microbial and human heat shock protein 60/65 in atherosclerosis. Arterioscler Thromb Vasc Biol. 23:1060–1065. 2003.
Peveri P., Walz A., Dewald B., Baggiolini M. A novel neutrophil-activating factor produced by human mononuclear phagocytes. J Exp Med. 167:1547–1559. 1988.
Pockley AG. Heat shock proteins as regulators of the immune response. Lancet. 362:469–476. 2003.
Pockley AG., Georgiades A., Thulin T., de Faire U., Frostegard J. Serum heat shock protein 70 levels predict the development of atherosclerosis in subjects with established hypertension. Hypertension. 42:235–238. 2003.
Rus HG., Vlaicu R., Niculescu F. Interleukin-6 and interleukin-8 protein and gene expression in human arterial atherosclerotic wall. Atherosclerosis. 127:263–271. 1996.
Schroder JM., Christophers E. Secretion of novel and homologous neutrophil-activating peptides by LPS-stimulated human endothelial cells. J Immunol. 142:244–251. 1989.
Srivastava P. Roles of heat-shock proteins in innate and adaptive immunity. Nat Rev Immunol. 2:185–194. 2002.
Steinberg D., Parthasarathy S., Carew TE., Khoo JC., Witztum JL. Beyond cholesterol. Modifications of low-density lipoprotein that increase its atherogenicity. N Engl J Med. 320:915–924. 1989.
Tedgui A., Mallat Z. Cytokines in atherosclerosis: pathogenic and regulatory pathways. Physiol Rev. 86:515–581. 2006.
Wang JM., Sica A., Peri G., Walter S., Padura IM., Libby P., Ceska M., Lindley I., Colotta F. Mantovani A. Expression of monocyte chemotactic protein and interleukin-8 by cytokine-activated human vascular smooth muscle cells. Arterioscler Thromb. 11:1166–1174. 1991.
Whitley D., Goldberg SP., Jordan WD. Heat shock proteins: a review of the molecular chaperones. J Vasc Surg. 29:748–751. 1999.
Wick G., Perschinka H., Millonig G. Atherosclerosis as an autoimmune disease: an update. Trends Immunol. 22:665–669. 2001.
Wu YM., Robinson DR., Kung HJ. Signal pathways in up-regulation of chemokines by tyrosine kinase MER/NYK in prostate cancer cells. Cancer Res. 64:7311–7320. 2004.
Yang X., Coriolan D., Murthy V., Schultz K., Golenbock DT., Beasley D. Proinflammatory phenotype of vascular smooth muscle cells: role of efficient Toll-like receptor 4 signaling. Am J Physiol Heart Circ Physiol. 289:H1069–1076. 2005.
Yang X., Murthy V., Schultz K., Tatro JB., Fitzgerald KA., Beasley D. Toll-like receptor 3 signaling evokes a proinflammatory and proliferative phenotype in human vascular smooth muscle cells. Am J Physiol Heart Circ Physiol. 291:H2334–2343. 2006.
Yue TL., Wang X., Sung CP., Olson B., McKenna PJ., Gu JL., Feuerstein GZ. Interleukin-8. A mitogen and chemoattractant for vascular smooth muscle cells. Circ Res. 75:1–7. 1994.
Go to :
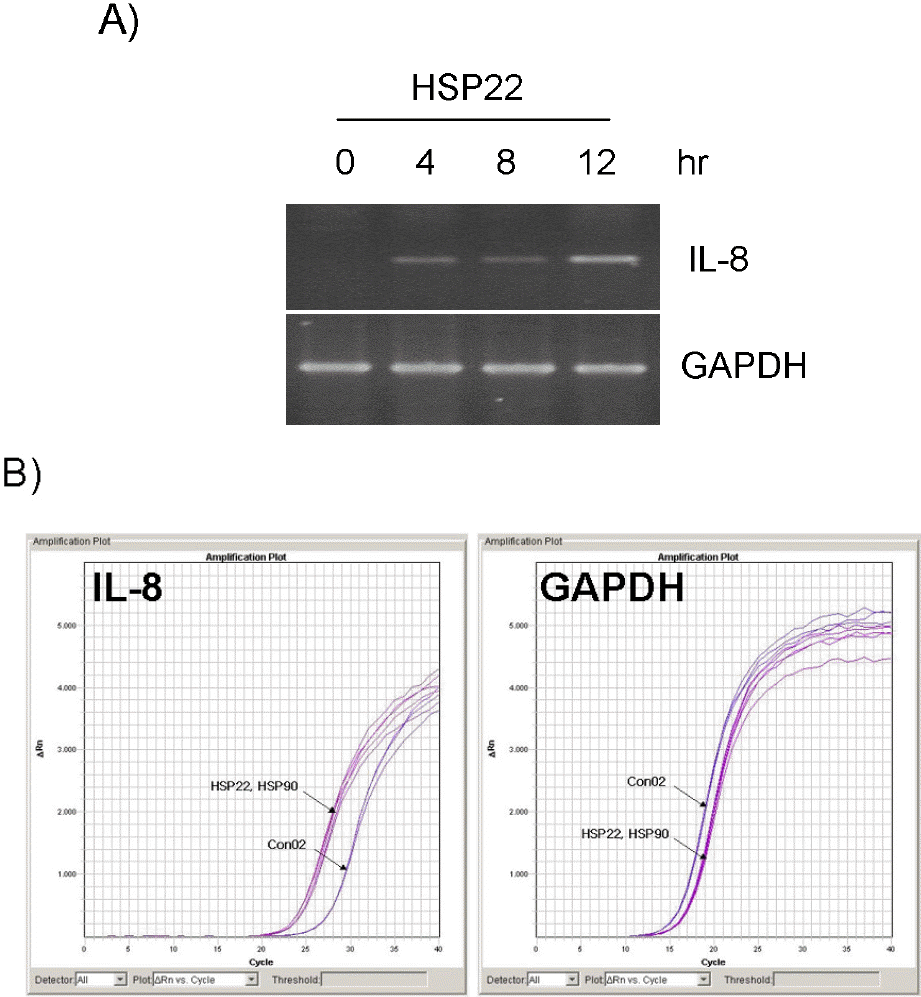 | Fig. 1.Induction of IL-8 in VSMCs by HSP22. (A) AoSMCs were treated with 100 ng/ml HSP22 for indicated periods, and IL-8 transcript was amplified by RT-PCR. (B) IL-8 transcript was quantified by quantitative real-time PCR with BSA a control after AoSMCs were treated with 100 ng/ml of HSP22, HSP90 and BSA (con02) for 12 h. |
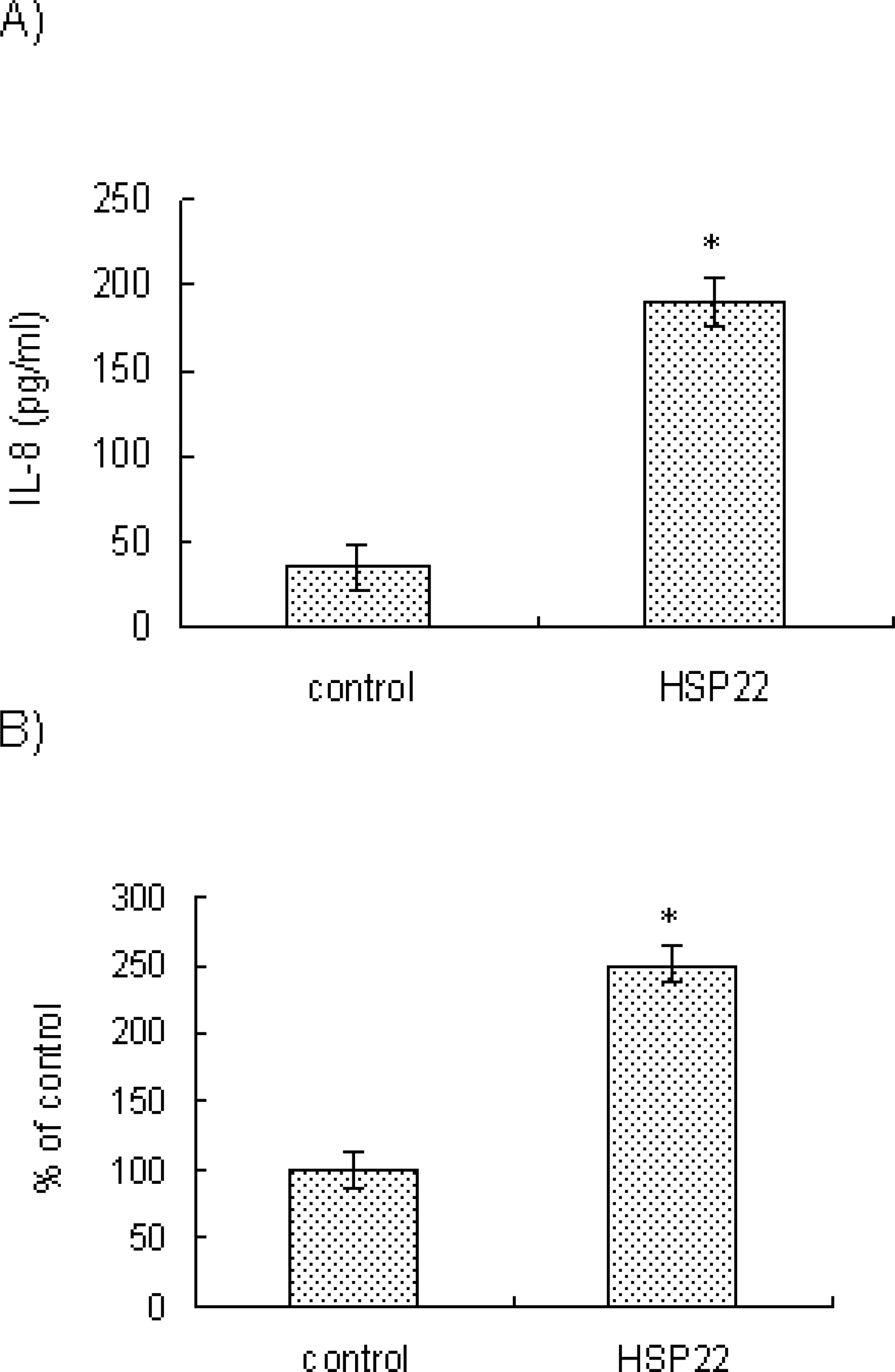 | Fig. 2.Effects of HSP22 on IL-8 release and IL-8 promoter activity in VSMCs. (A) AoSMCs (1×106 cells/100-mm culture dish) cultured in growth media were incubated for 12 h in the presence of BSA (control) or HSP22 (100 ng/ml). The culture media were harvested. IL-8 secreted in the culture medium was measured by ELISA. Data are expressed as mean±SD (n=3 replicates/group). ∗p<0.01 vs. control. (B) A7r5 cells were transfected with the wild-type pIL-8-Luc651 construct. The transfected cells were incubated in the presence of BSA (control) or HSP22 (100 ng/ml, for 8 h), and processed for luciferase and β-galactosidase assays. Luciferase activity (×106) was normalized to β-galactosidase activity. Induction was calculated relative to the activity of control cells. Data are expressed as mean±SD (n=3 replicates/group). ∗p <0.01 vs. control. |
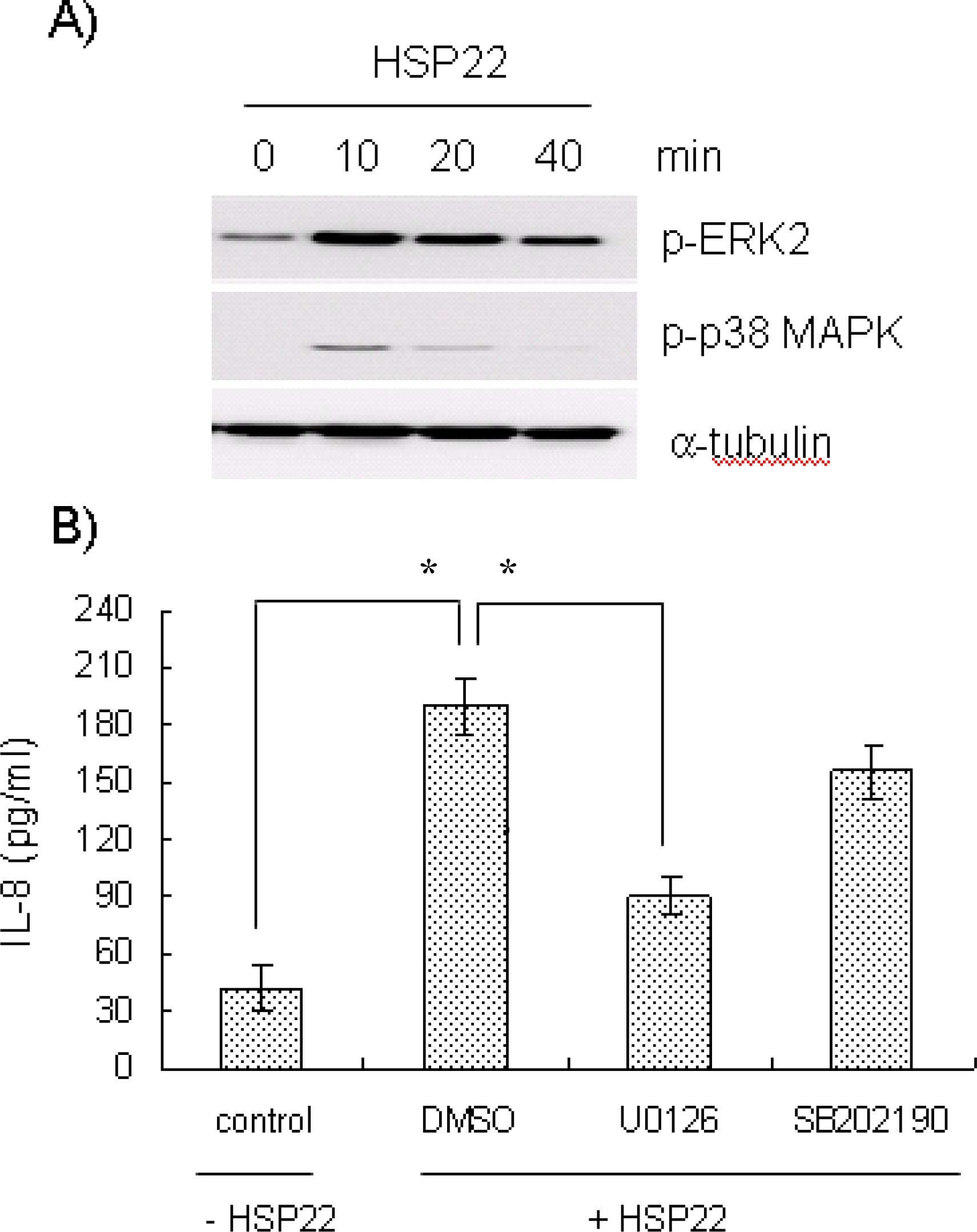 | Fig. 3.Effects of MAPKs inhibitors on HSP22-induced IL-8 release. (A) Human aorta smooth muscle cells were exposed to HSP22 (100 ng/ml) for the indicated time periods, and cell lysates were then prepared. An equal amount of protein was subjected to Western blot analysis with antibodies for α-tubulin, phospho-ERK2 (p-ERK2), phospho-p38 MAPK (p-p38 MAPK) and α-tubulin. (B) AoSMCs were pre-treated with U0126 or SB202190 (10 μM each, for 2 h) and stimulated with (+) or without (–) HSP22 (100 ng/ml, for 12 h). Secreted IL-8 was measured by ELISA. Data are expressed as mean±SD (n=3 replicates/group). ∗p<0.01. |
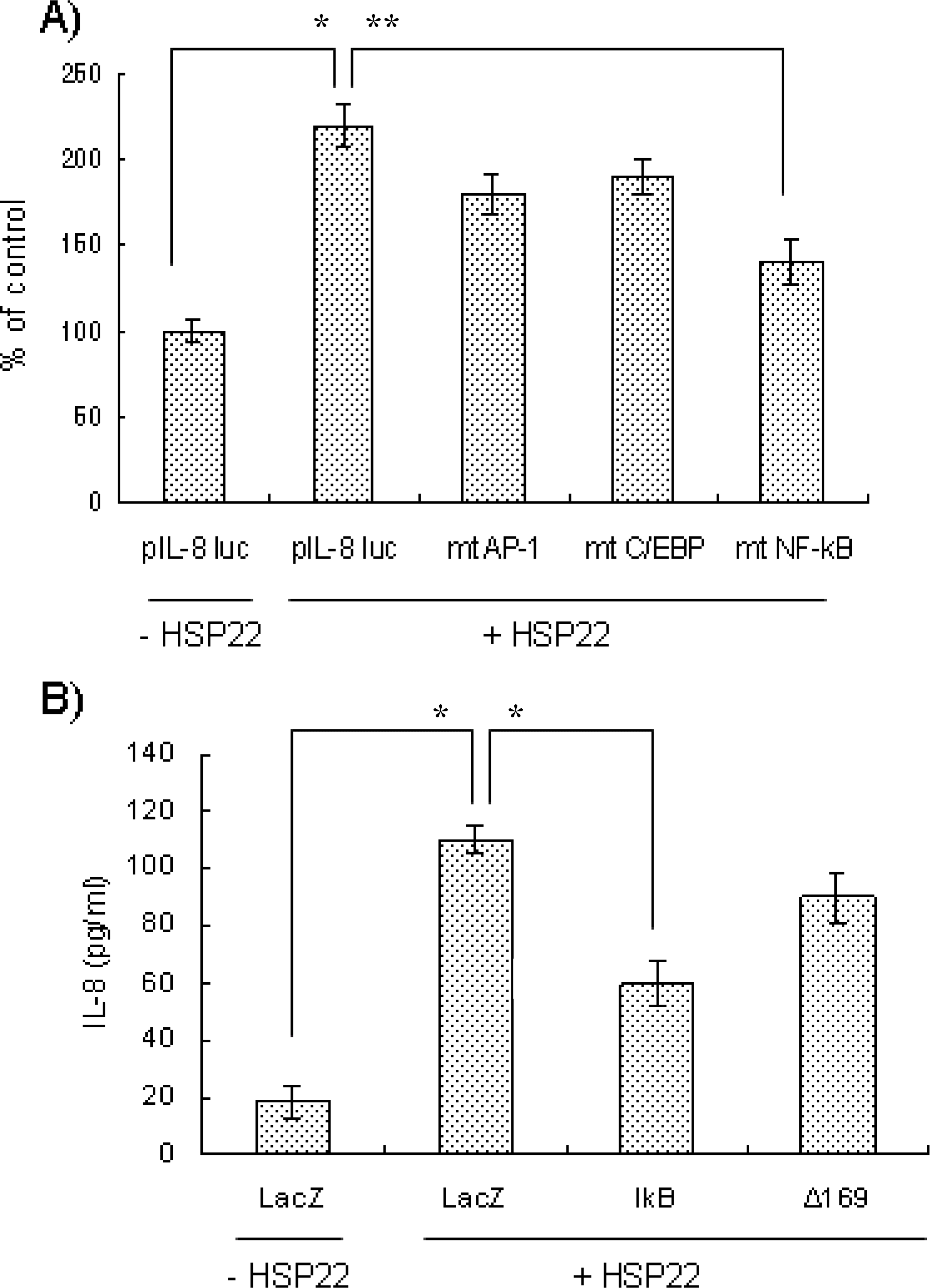 | Fig. 4.Effects of transcriptional elements on HSP22-induced IL-8 up-regulation. (A) A7r5 cells were transfected with the wild-type pIL-8-Luc651 construct or the indicated mutant construct. The transfected cells were stimulated with (+) or without (–) HSP22 (100 ng/ml) for 8 h and processed for luciferase and β-galactosidase assays. Induction was calculated relative to the activity of cells incubated in the absence of HSP22. Data are expressed as mean±SD (n=3 replicates/group). ∗p<0.01, ∗∗p<0.05. (B) AoSMCs were infected with indicated recombinant adenoviruses (MOI: 100) and stimulated with (+) or without (–) HSP22 (100 ng/ml, for 12 h). The amount of secreted IL-8 was measured by ELISA. Data are expressed as mean±SD (n=3 replicates/group). ∗p<0.01. |
 XML Download
XML Download