

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Glaucoma is a progressive optic neuropathy caused by death of the retinal ganglion cells and is the leading cause of irreversible blindness worldwide. The mechanism by which this progressive retinal ganglion cell death occurs is not fully understood. It is clear that multiple causes can give rise to the common effect of ganglion cell death. The ciliary secretion of aqueous humor usually remains normal in glaucoma; therefore, it is thought that impaired drainage through the trabecular pathway caused by increased resistance is the primary cause of increased intraocular pressure (IOP) in primary open-angle glaucoma [12]. While most glaucoma medications approved for clinical use act either on the uveoscleral pathway and/or aqueous humor formation, none target the major site of resistance to aqueous humor outflow in glaucoma, namely, the trabecular meshwork/Schlemm's canal (TM/SC; a conventional outflow pathway). Therefore, novel pharmacological targets that can affect the conventional outflow pathway are highly desirable. Recently, several new drugs targeting the trabecular outflow pathway have entered clinical development [3], such as Rho-associated kinase (ROCK) inhibitors, adenosine agonist, and statin [4].
Rho GTPase activity is regulated by physiological ligands and growth factors that engage membrane bound receptors; upon conversion to its GTP-bound (activated) form, Rho GTPase then stimulates Rho kinase activity. Inhibition of Rho GTPase activity in TM and SC cells induces changes in cell shape and leads to decreases in actin stress f ibers and cell adhesion and in myosin light-chain (MLC) phosphorylation [5]. ROCK inhibitors Y-27632, H-1152, and fasudil have been demonstrated to induce very rapid and long-lasting decreases in IOP [6789]. Adenosine agonists increase trabecular out flow by changing TM cell volume and ion transport and by remodeling the extracellular matrix (ECM) [10]. Cholesterol-lowering statin significantly increased aqueous outf low facility in an organ- cultured porcine anterior eye segment perfusion model [11], and it has been reported that human patients who were administered statin drugs had a reduced risk of glaucoma [12].
Free radical nitric oxide (NO) has multiple functions in different cells. TM cells have shown smooth muscle cell-like activity in morphological and electrophysiological studies [1314]. NO increases aqueous outflow by relaxing TM, and levels of NO and nitric oxide synthase (NOS) expression are reduced in glaucomatous human eyes [151617]. Although it is known that ROCK inhibitor and statin increase the expression of endothelial nitric oxide synthase (eNOS) [1819], their effects on the expression of eNOS in human TM cells are still unknown.
The significance of matrix metalloproteinases (MMPs) in outflow resistance was shown when treatment of anterior segments in perfusion culture with MMPs was found to increase outflow, while specific inhibition of MMP activity decreased outflow facility [20]. Although ROCK inhibitors do not affect the expression of MMP-2, which are involved in the remodeling of ECM [21], treatment with adenosine significantly increases its expression [22], and the effect of statin on its expression has not been studied in TM cells.
This study was performed to compare the effects of ROCK inhibitors, adenosine, or statin on the in vitro trabecular outflow by measuring the barrier function of a TM cell monolayer. The effects of these trabecular outflow drugs on the production of NO a nd expression of eNOS and MMP-2 mRNA were also studied.
Materials and Methods
Cell culture and experimental treatment
This study followed the tenets of the Declaration of Helsinki and was approved by the institutional review board/ethics committee of Daegu Catholic University Hospital (No. CR-16-117-L).
TM cell cultures were established f rom a n enucleated human eye obtained from the eye bank and transported on ice within 2 hours after exsanguination, as previously described [23]. Briefly, TM tissues were excised by dissecting a continuous strand of tissue between the line of Schwalbe and the scleral spur by teasing away the TM tissue using a curette. The excised TM tissues were placed in a sterile culture dish with Dulbecco's modified Eagle's medium (DMEM) containing 15% fetal bovine serum (FBS), 2 mM glutamine, 50 µg/mL gentamicin, and 2.5 mg/mL fungizone and left undisturbed for 3 to 5 days in a 37℃ incubator with a 5% CO2 atmosphere. After observation of initial cell growth, the explants were removed, and the cultures were maintained in media containing 10% FBS.
Cultures approaching confluency were trypsinized and inoculated into culture plates. After attachment, the cells were washed three times with serum-free medium and then cultured for 24 hours in media supplemented with 10% FBS containing the following drugs. ROCK inhibitor (10 µM or 25 µM Y-27632; Sigma, St. Louis, MO, USA), adenosine A1 agonist (0.1 µM or 1 µM N6-cyclohexyladenosine [CHA], Sigma), or statin (15 or 30 µM simvastatin, Sigma) was added to the cultures, as in previous studies [71922].
MTT (3-[4, 5–dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide) assay
Cell survival was determined by a rapid MTT colorimetric assay. The MTT assay is based on the tetrazolium salt MTT that reacts with living but not dead cells. As signals are generated, the optical density is directly proportional to the number of cells [23]. For the assay, 100 µL of a MTT stock solution (5 mg MTT/mL phosphate buffered saline) was added to each well and incubated for 4 hours at 37℃, after which all media was removed from the well. After 0.5 mL of dimethyl sulfoxide was added to each well, 100 µL of solution from each well was t ransferred t o a 96-well plate and analyzed on a multi-well scanning spectrophotometer (λ = 570 nm, Fluostar Optima; BMG Labtech, Offenburg, Germany).
Measurement of monolayer cell permeability with carboxyfluorescein
A permeability study of the TM cell monolayer was performed as previously described with minor modification [24]. Briefly, primary cultured human TM cells were incubated in the inner chamber (insert diameter, 12 mm; pore size, 0.4 mm) of a 12-well plate (No. 3460, Transwell; Corning, Tewksbury, MA, USA) as 2×104 cells/mL supplemented with 10% FBS. After the cells reached confluency, the media was changed to 1% serum-containing DMEM to avoid the effects of growth factors and proteins in serum; then, the TM cells were exposed to each drug for 24 hours. After washing three times with PBS, 50 µM of the tracer carboxyfluorescein (Sigma-Aldrich, St. Louis, MO, USA) was added to each well. The media was collected from the outer well to analyze fluorescence after 2 hours, and the concentration of carboxyfluorescein in the collected media was measured using a spectrofluorometer (Fluostar Optima, BMG Labtech) with an excitation wavelength of 490 nm and an emission wavelength of 530 nm.
Measurement of monolayer trans-endothelial electron resistance
TM cells were seeded at 2×104 cells/well and were grown until confluent in the inner well (pore size, 0.4 mm; diameter, 12 mm) in 12-well culture plates (Transwell, Corning) [7,25]. Twenty-four hours after exposure to each drug, trans-endothelial electron resistance (TEER) was measured with an electrical resistance system (epithelial voltohmmeter, EVOM2; World Precision Instruments, Sarasota, FL, USA) according to the manufacturer's instructions, and the results were recorded as net value (Ω cm2).
Measurement of NO production
Nitrite concentrations in the media were measured using the Griess reaction [26]. Briefly, media samples were collected from each well following appropriate treatment and reacted with modified Griess reagent by mixing equal volumes at room temperature for 15 minutes. Optical density was then measured on a multi-well scanning spectrophotometer at 540 nm. The nitrite concentration was determined f rom a comparison of a bsorbance with that of a standard solution of sodium nitrite in media. The background absorbance, measured using the media alone, was subtracted from all values.
RT-PCR for measurement of the expression of eNOS and MMP-2 mRNA
After exposure to each drug, total RNA was extracted with Trizol (Invitrogen, Carlsbad, CA, USA). An RNA denaturation mix consisting of isolated RNA, oligo dT primers, and nuclease-free water was denatured. RT-PCR was performed using oligonucleotide primers specific to eNOS and MMP-2. Two tubes were run in parallel with the second tube containing only Platinum Taq polymerase to assure that the source of the RT-PCR product was mRNA. cDNA was synthesized by adding prime RT premix. Taq Green Master Mix and 10 pM each of forward and reverse primers (eNOS: forward primer, ctg gct ttc cct tcc agt tc, 225 bp; reverse primer, cct tcc aga tta agg cgg ac, 225 bp) (MMP-2: forward primer, ctc gtg cct tcct aag tct gg, 251 bp; reverse primer, ggc gtt ccc ata ctt cac ac, 251 bp) were added to the synthesized cDNA. The amplification reaction was carried out for 30 cycles on a DNA Engine Cycler (Bio-Rad, Hercules, CA, USA). The amplified PCR products were analyzed using Multi-gauge software (Fujifilm, Tokyo, Japan) after electrophoresis. β-actin was used as an internal standard.
Statistical analysis
Every experiment was performed in triplicate, and the data are expressed as mean ± standard error of the mean of the number of wells analyzed. Experimental differences between the results of control cultures and a single treatment group were evaluated using Student's t-test. A p-value less than 0.05 was considered statistically significant.
Results
Cell culture
The TM cells started to grow into the surrounding tissue explants as a monolayer after 2 or 3 weeks of primary culture. The presence of TM cells was confirmed by their characteristic morphology and characteristic growth pattern as satellite colonies distant from the tissue explants [27].
Effects of trabecular outflow drugs on TM cell survival and NO production
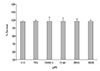
Treatment with each drug tested did not significantly inhibit the survival of TM cells (p > 0.05) (Fig. 1). Thus, the drugs did not affect cell survival and proliferation.
Effects of trabecular outflow drugs on the NO production and eNOS mRNA expression
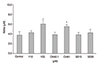
After 24 hours of exposure, treatment with 25 µM Y-26732 or 1.0 µM CHA significantly increased the nitrite concentration in the media in a dose-dependent manner (p = 0.035, p = 0.043) (Fig. 2). Treatment with each drug also significantly increased the expression of eNOS mRNA (p < 0.05)(Fig. 3). Treatment with Y-26732 increased the eNOS mRNA expression to a level higher than that produced by CHA or simvastatin.
Effects of trabecular outflow drugs on barrier function
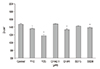
The permeability assay using a flux of carboxyfluorescein showed that treatment with each drug tested except 0.1 µM CHA increased the concentration of carboxyfluorescein in the outer well in a dose-dependent manner compared to non-exposed controls (p < 0.05) (Fig. 4). Treatment with 10 µM or 25 µM Y-26732 produced 12.74% and 21.25% higher concentrations of carboxyfluorescein, respectively, in a dose-dependent fashion compared to treatment with the other drugs.
The TEER of the TM cell monolayers was significantly lower after adding 10 µM or 25 µM Y-27632, 1.0 µM CHA, or 30 µM simvastatin compared with that of controls (p < 0.05) (Fig. 5). Treatment with 25 µM Y-26732 produced a significantly greater 28.91 Ω cm2 decrease in TEER compared to treatment with the other drugs.
Effects of trabecular outflow drugs on the expression of MMP-2 mRNA
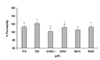
Treatment of the TM cells with 0.1 µM or 1.0 µM CHA resulted in an increase in the level of MMP-2 mRNA up to 11.98% or 32.61%, respectively (p = 0.012, p = 0.001) (Fig. 6). On the contrary, treatment of the TM cells with Y-27632 did not affect MMP-2 mRNA expression, while the addition of simvastatin inhibited its expression.
Discussion
The TM/SC complex is considered the conventional aqueous humor outflow pathway and is a primary site of increased resistance to aqueous humor outflow in glaucoma. There is a significant need for glaucoma drugs that specifically target the physiologic cause of elevated IOP and thereby enhance trabecular outflow. The most widely prescribed glaucoma drugs, prostaglandin analogues, lower IOP by increasing the aqueous drainage through the unconventional uveoscleral outf low pathway. The older, non-prostaglandin drug classes, β-blockers, α-agonists, and carbonic anhydrase inhibitors, lower IOP by decreasing the production of aqueous humor. Recently, several new classes of drugs targeting the trabecular outflow pathway have been introduced, and their diverse effects are under investigation [4].
Rho kinase has been suggested as a potential therapeutic target for the development of drugs to modulate IOP in glaucoma patients. Considering ROCK inhibitor-induced changes in MLC phosphorylation and actin-myosin organization, it seems that cellular relaxation and loss of cell-substratum adhesions in TM and SC cells could result in either increased paracellular fluid flow across the SC or an altered flow pathway through the juxtacanalicular tissue, thereby lowering resistance to outflow [72428].
The statin drugs induce changes in cell shape and actin cytoskeletal organization and decrease MLC phosphorylation in TM cells, all of which are events that are likely to lead to cellular and tissue relaxation. In addition, these effects of statins appear to be mediated by inhibition of isoprenylation of the small GTP-binding proteins such as Rho-GTPase [11]. Compared to ROCK inhibitors or statin, adenosine increases trabecular outflow by inducing ECM change [10]. Adenosine A1 receptor exist in TM cells, and CHA modulates MMP-2 secretion in TM cells [222930].
It is known that ROCK inhibitor, adenosine, and statin all increase trabecular outflow, although no comparative study of their effects on the increase in trabecular outflow had been reported. Thus, this study was performed to compare their effects on in vitro trabecular outflow by measuring the permeability and resistance of TM cell monolayers. The results showed that treatment with each drug tested in the present study increased permeability, except 0.1 µM CHA. Treatment with the ROCK inhibitor Y-26732 more greatly increased the permeability compared to adenosine or statin. Furthermore, treatment with 25 µM Y-26732 most significantly lowered the TEER of TM cell monolayer.
NO is involved in the regulation of trabecular outflow, and external administration of NO results in decreased IOP [31]. There has been evidence that treatment with ROCK inhibitor or statin increased eNOS expression [1819], but their effects on the expression of eNOS in human TM cells had not yet been investigated. Moreover, adenosine's effects on the expression of eNOS and NO production were still not explored. In the present study, treatment with the ROCK inhibitor Y-26732 increased the expression of eNOS and NO production more significantly than adenosine or statin. Thus, increased NO production by ROCK inhibitor might have an additive effect on increasing outflow.
Treatment with CHA significantly increased MMP-2 expression. On the contrary, treatment with Y-27632 did not significantly increase MMP-2 expression, and treatment with simvastatin inhibited MMP-2 expression. Thus, it is likely that adenosine has another mechanism through which it increases trabecular outflow by remodeling of the ECM in addition to cellular morphological changes, as produced by ROCK inhibitor or statin. Treatment with statin had no effect on MMP-2 expression.
ROCK inhibitors show multiple effects other than decreasing IOP, such as antioxidant and anti-inflammatory activities [3233]. Furthermore, ROCK inhibitors have an antifibrotic effect and decrease scar formation after trabeculectomy [33].
Clinical trabecular outflow modulators based on preclinical efficacy models are currently being explored. Results of clinical trials of the ROCK inhibitor ripasudil (K-115) have been reported [3435]. The neuroprotective effect of another ROCK inhibitor, fasudil, has been evaluated [3637]. Among adenosine agonists, clinical trials on the favorable IOP-lowering effect of trabodenoson (INO-8875) has been reported recently [38]. These clinical studies revealed that ROCK inhibitors showed a stronger IOP-lowering effect than adenosine or statin, in agreement with our present in vitro study. However, there are limitations of these newly proposed mechanisms. For example, the incidence of ocular hyperemia with ROCK inhibitors is higher than that with latanoprost, while the magnitude of IOP lowering is not superior. Therefore, the risk to benefit ratio for patients seems unfavorable, and we must continue to identify new mechanisms through which to lower IOP [39].
In conclusion, ROCK inhibitor more highly increased the permeability and decreased the resistance of a TM cell monolayer than adenosine or statin. In addition, increased expression of eNOS mRNA and NO production by ROCK inhibitor might result in further IOP reduction. Thus, trabecular outflow might be more enhanced by ROCK inhibitor than by adenosine or statin. In the future, more clinical studies will be needed to explore and compare IOP-lowing effects in humans.


 XML Download
XML Download