

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Corneal dystrophies are hereditary diseases involving corneal opacities at different layers of the cornea. They are primary corneal lesions that are not associated with prior inflammation or trauma [1]. Macular corneal dystrophy (MCD) is an autosomal recessive disorder characterized by bilateral progressive stromal clouding and central corneal thinning [2]. It is the least common form of the classic stromal dystrophies in most countries, especially in East Asia [3,4]. MCD occurs in primarily the first decade of life, beginning with a fine superficial stromal haze in the central stroma. Gradually, the opacification extends and involves the entire cornea, resulting in visual impairment, which generally requires penetrating keratoplasty.
Here, we report a patient with MCD and decreased visual acuity, who underwent penetrating keratoplasty. We confirmed the diagnosis via histopathologic studies such as light microscopy, electron microscopy, and DNA analysis. We found a mutation of CHST6 gene mutation (c.613 C>T [p.Arg205Trp]), which has never been reported previously.
Case Report
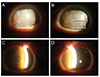
A 59 year-old female with a family history of MCD presented to our clinic with progressive loss of vision over a 40-year period. Her visual acuity was finger count/50 cm OU. Slit lamp examination revealed diffusely hazy corneas with bilateral opacities. There were multiple irregular grayish-white, dense, poorly delineated spots in the stroma. Because her parents died at an early age, her family history was unclear. Of note, her only son complained of foggy vision and was 37 years old. His slit lamp examination revealed a similar, but less severe appearing corneal exam compared to that of his mother (Fig. 1).
Penetrating keratoplasty was performed on the right eye. A 7.75 mm donor button was sutured into a 7.5 mm host bed. Histopathologic examinations were conducted on the excised corneal buttons obtained from the procedure. Genomic DNA was extracted from the leukocytes of her peripheral blood via standard procedures. One year post-operatively, that patient had a best-corrected visual acuity of 20 / 100 OD and a clear cornea graft.
Informed consent for both the clinical examinations and DNA analyses was obtained from the patient and her son in accordance with the Declaration of Helsinki. The study was approved by the institutional review board at the Catholic University of Korea, St. Mary's Hospital.
Histopathologic examination
Hematoxylin and eosin (HE), alcian blue, periodic acid-Schiff (PAS), colloidal iron, and Masson's trichrome stains were performed on the specimen using standard techniques. For transmission electron microscopy, the corneal specimen was immersed immediately in a fixative solution containing 3% glutaraldehyde with 0.2 M sodium cacodylate at a pH of 7.4. After overnight fixation, the fixative solution was removed and replaced with a phosphate buffer, followed by 1% osmium tetroxide buffered with sodium cacodylate. After one hour, the osmium was replaced with increasing concentrations of ethanol through propylene oxide, and the tissue was embedded in the epoxy. The embedded tissue was then sectioned with an ultra-microtome into 1 µm-thick sections, and stained with toluidine blue. The area to be observed was placed under a light microscope, ultra-sectioned from 60 to 100 nm in thickness, double stained with uranyl acetate and lead citrate, and examined via transmission electron microscope (JEM 1010; JEOL, Tokyo, Japan).
DNA analysis
The unique exon involving the coding region, exon 3, was amplified via polymerase chain reaction (PCR) with the previously described primers [5]. Thermal cycling was conducted via the following protocol. The cycling program began with an initial denaturing step of 5 minutes at 95℃ followed by 33 cycles of 94℃ for 30 seconds, 53℃ to 57℃ for 30 seconds, and 72℃ for 45 seconds with a final extension step at 72℃ for 10 minutes. The PCR products were purified and sequenced directly on both strands using an automatic DNA sequencer (ABIPrism 377XL; Applied Biosystems, Foster City, CA, USA). The nucleotide sequences were compared with the published cDNA sequence of CHST6 (NM_021615) [6].
Results
Light microscopy showed a normal epithelium and Bowman's membrane. HE staining revealed faintly basophilic deposits between the stromal lamellae, and within keratocytes and endothelial cells. These deposits were positive to alcian blue, PAS, and colloidal iron stain, but negative to Masson's trichrome stain (Fig. 2). Electron microscopy revealed keratocytes distended by membrane-bound intracytoplasmic vacuoles containing electron-dense fibrillogranular material. These vacuoles harbored dense osmophilic whorls. Similar vacuoles were also present in the interstromal lamellae and endothelial cells (Fig. 3).
Analysis of the entire CHST6 coding region revealed distinct genetic defects. One missense mutation was identified in a homozygous state (p.Arg205Trp [c.613C>T]), which had not been previously reported. Samples from the patient's son also showed the same missense mutation (p.Arg205Trp [c.613C>T]) (Fig. 4).
Discussion
In this study, we describe histopathological findings and a novel mutation in the CHST6 gene (p.Arg205Trp [c.613C>T]) in a case of MCD [7]. Light microscopy revealed abnormal deposits of glycosaminoglycans in Bowman's histiocytes and keratocytes, as well as between the stromal lamellae, Descemet's membrane, and endothelium. These glycosaminoglycans stained positively with alcian blue, colloidal iron, and PAS [8]. Electron microscopy revealed that these deposits corresponded to electron-lucent fibrillogranular material visible within membrane-bound intracytoplasmic vacuoles [9]. Such abnormalities have been identified as sequelae of an error in glycosaminoglycans metabolism within the cornea. In particular, errors in proteoglycan keratan sulfate (KS) metabolism have resulted in abnormal intra- and extracellular deposition [10]. The product of the CHST6 gene, corneal N-acetylglucosamine-6-sulfotransferase (C-GlcNac6ST), has been demonstrated to catalyze the sulfation of GlcNAc in KS. In normal corneal tissue, KS is an important glycosaminoglycan, which exists in a highly sulfated form. KS performs a crucial role related to the structure and transparency of the cornea. CHST6 can transfer a sulfuric acid group from the 3'-adenosine 5'-phosphate acid to KS via competition with an endogenous or exogenous substrate [11]. The variations in the coding region of CHST6 may reduce enzyme activity or cause it to be lost, resulting in a low sulfated form or non-sulfated form of KS. Due to the loss of its soluble properties, non-sulfated KS cannot be completely metabolized, inducing the deposition of sediment in the corneal stroma [12].
The mutation identified in this Korean patient is different from those reported previously in Asians (Japanese and Chinese) or whites (American, British, and Icelandic) [5,6,13,14]. Although the p.Arg205Gln mutation has been reported in a Southern Indian patient, the p.Arg205Trp mutation has not been reported previously [15]. The immunophenotype of MCD was not performed in our study since we could not test KS serum levels. Regardless of the type of macular corneal dystrophy, the patient showed indistinguishable clinical characteristics linked to the same responsible gene.
In summary, we identified a novel homozygous missense mutation in CHST6 in a Korean patient with macular corneal dystrophy. This novel gene mutation expands the mutation spectrum of the CHST6 gene and contributes to the study of molecular pathogenesis of corneal dystrophy. Further studies on these mutations would be helpful in further understanding the molecular mechanisms underlying macular corneal dystrophy.



 XML Download
XML Download