

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Muir-Torre syndrome (MTS) is a clinical variant of hereditary non-polyposis colorectal cancer (Lynch syndrome), and is defined as an autosomal dominant condition with simultaneous sebaceous neoplasms of the skin and visceral malignant disease resulting from germline mutations in DNA mismatch repair (MMR) genes [1]. This is a rare disease and the most common skin tumors associated with it are sebaceous adenomas, carcinomas, keratoacanthomas, and basal-cell carcinomas with sebaceous differentiation [2]. The most common visceral malignancy is colorectal cancer. Although the exact incidence is unclear, there are only a few reports of a combination of eyelid tumors and breast cancer, and reports of MTS in Asian populations are rare. Here, we report a case of MTS that presented with multiple cancers of both eyelids and the breast with lack of mutS homolog (MSH) 2 staining.
Case Report
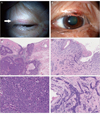
A 71-year-old woman presented with a swelling on her left upper eyelid in 2009. She also had a small mass on her right upper eyelid that been present for the previous 3 years. She had a history of breast cancer occurring in 1991, which was treated successfully without recurrence for 18 years. Laboratory data tests showed no remarkable changes. There was a significant family history of cancer; two of her brothers had also been diagnosed with cancers. Surgical excision and skin biopsy identified a sebaceous carcinoma in her right eyelid and a morphea-like basal cell carcinoma of her left eyelid (Fig. 1). Immunohistochemical analysis was done using anti-mutL homolog (MLH) 1 (mouse monoclonal; 1:50; BD Pharmingen, San Diego, CA, USA) and anti-MSH2 (mouse monoclonal; 1:40; Calbiochem, San Diego, CA, USA). A MLH1 gene product was present in both tumors, but no MSH2 genes were found in either tumor (Fig. 2). Colon cancer tissue obtained from another patient was used as a positive control specimen, with the patient's permission.
Discussion
MTS is a rare autosomal dominant genodermatosis with variable expressivity, and is regarded as a variant of Lynch syndrome [3,4]. There is little or no information about the main epidemiological features [5]. Inherited colorectal cancer syndromes such as familial adenomatous polyposis, mutY human homologue (MYH)-associated polyposis, and Lynch syndrome/hereditary nonpolyposis colon cancer are less common but account for as much as 5% of colorectal cancer cases [6]. A diagnosis is made based on the presence of at least one sebaceous neoplasm associated with visceral malignancy, or alternatively, multiple keratoacanthomas with visceral malignancies and a family history of MTS. Fifty-six percent of the skin lesions in MTS develop after diagnosis of the first internal malignant disease, 6% appear concomitantly, and 22% occur as the first tumor of the syndrome [7]. The most common visceral malignancies in MTS are colorectal carcinomas. Other types of cancer, such as breast cancer, are uncommon (4%) [2]. The present case had a history of breast cancer, and it was possible that her eyelid neoplasms resulted from breast cancer metastasis. However, this is unlikely because the patient's breast cancer did not recur for a period of 18 years and 2 different types of neoplasms were found in the eyelids. More importantly, immunohistochemical staining for hMSH2 and hMLH1 was consistent with a diagnosis of MTS, in addition to clinical findings that met the clinical criteria for MTS [4,8,9]. Considering these results, it would be safe to speculate that the case belonged to a spectrum of MTS conditions.
Sebaceous gland tumors are much more common in Asians than in Caucasians, with a reported frequency of 10.2% to 32.7% of malignant tumors in Eastern Asians in comparison to 0% to 3.2% in the US and Europe [10]. Indeed, sebaceous gland tumors in Caucasians are so rare that their presence is considered a marker for MTS, and lead to a search for an occult malignancy [11]. The incidence of MTS in the Asian population is not clear, but it is likely to be underestimated. Because sebaceous gland neoplasms of the eyelid are so common in Asia, and most of them are successfully treated by local excision, their presence is not regarded as a specific event. Even if another visceral malignancy occurred in the same patient after a long interval, it would often be diagnosed as a coincidence of independent tumors. In fact, the diagnosis would not have been easy without the help of immunohistochemical analysis for MLH1 and MSH2, which were used in the present case.
Many MTS patients demonstrate germ-line mutations in genes encoding DNA MMR proteins such as MLH1 or MSH2 and, less commonly, MSH6. These proteins function to detect and repair errors in base pairing that occur during DNA replication, especially in regions of DNA characterized by repetitive mono- or dinucleotide repeats, termed microsatellites. Conceptually, MMR genes can be considered tumor suppressor genes. MMR protein mutations, when paired with a second somatic mutational hit of the remaining functional allele, leads to increased susceptibility to tumor formation [1]. Immunohistochemistry assessing MSH2, MLH1, and MSH6 activity, and microsatellite instability (MSI) analysis are currently the two main screening tests for MTS. MMR genes encode proteins that recognize and remove improper genomic insertions and deletions and nucleotide mismatches that arise during DNA replication and recombination. Collectively, these genes maintain the integrity of the genome and prevent mutations from accumulating. In MTS, individuals are born with one defective copy of an MMR gene and acquire a second somatic mutation in the other allele during their lifetimes [8]. The first mutations identified in MTS involved the MMR genes MSH2 and MLH1, the same genes known to cause Lynch syndrome. Recent advances in immunohistochemistry and MSI analysis have made the diagnosis of MTS much easier and more reliable. MSI analyses and reviews of family history should guide cancer surveillance options for patients and their extended families after immunohistochemistry.
Patients with visceral malignant disease as part of MTS tend to survive longer than those with visceral malignant diseases outside of the syndrome, and even metastatic disease can respond to aggressive surgical treatment [5]. The role of ophthalmologists is much larger than before because 22% of skin lesions in MTS develop prior to the diagnosis of the first internal malignant disease [3]. Generally, sebaceous skin tumors characteristic of MTS can be regarded as premonitory physical characteristics for an underlying cancer predisposition [5]. At present, immunohistochemical studies can be easily carried out in most hospitals at low costs. Screening studies for MSH1 and MLH2 on eyelid tumors would give us important information about future treatment and follow-up programs for patients.

 XML Download
XML Download