

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Goldmann-Favre syndrome is an autosomal recessive hereditary vitreo-retinal degeneration characterized by impaired visual acuity, nyctalopia, vitreous degeneration, atypical peripheral pigmentary dystrophy, and peripheral and macular retinoschisis [1-3]. Typically, macular retinoschisis has a characteristic microcystic appearance on ophthalmoscopy, even without fluorescein staining on angiography [4]. Macular retinoschisis is allelic with enhanced S cone syndrome. Both conditions are caused by mutations in the nuclear receptor gene NR2E3 on chromosome 15 [5]. Manifestation of the mutations, however, may not be detected in all cases.
Case Reports
Case 1
A 21-year-old male presented with a progressive decrease in vision that had begun at the age of 8 years. His brother was known to have the same problem. The patient's best corrected visual acuity, with +4.00 DS / -0.50 DC × 180, was counting fingers at 50 cm in the right eye, and, with +1.00 DS / -1.50 DC × 20, was 6 / 24 in the left eye. The anterior segment examination was unremarkable. Indirect ophthalmoscopy revealed multiple vitreous membranes, peripheral vitreous detachment, peripheral retinal epithelial alterations, pigment clumping, equatorial chorioretinal atrophy inferior to the macula, lamellar macular holes, and foveal microcystic spaces in the patient's right eye. Lattice degeneration at the 6 o'clock position was also observed in the left eye. A diagnosis of Goldmann-Favre syndrome was made.
Time domain (Stratus) optical coherence tomography demonstrated confluent macular cystoid changes, as well as foveal retinoschisis in both eyes, with macular holes in the right eye. During electroretinogram evaluation, isolated rod responses were unrecordable for the right eye, and showed delayed implicit time with grossly reduced amplitude in the left eye. Maximal combined responses showed a negative b-wave. Oscillatory potentials were flat in both eyes. Isolated cone responses showed delayed implicit time with reduced amplitudes in both eyes. There was a gross delay in implicit time in both eyes with 30 Hz flicker. Fundus fluorescein angiography (FFA) revealed window defects corresponding to areas of atrophy of the retinal pigment epithelium (RPE). The patient underwent laser photocoagulation to the lattice in his left eye.
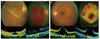
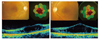
The patient returned for a follow-up visit 18 months later. His visual acuity was stable. Cataract formation was noted in his right eye. He had developed a bicycle wheel pattern of foveal schisis in his left eye. Imaging with spectral domain optical coherence tomography (SD-OCT; Copernicus, Optopol Technologies, Zawierci, Poland) of the right eye revealed lamellar macular holes with macular schisis, microcystic spaces, and vitreo-macular traction. SD-OCT imaging of the left eye revealed cystoid macular edema with inner layer schisis. Foveal thickness was 77 microns in the right eye and 672 microns in the left eye. Microperimetry (Nidek Technologies, Padova, Italy) showed a reduced mean retinal sensitivity of 0.0 db in the right eye and 5.5 db in the left eye, with a central dense scotoma in both eyes (Fig. 1).
Pedigree construction and venous blood sampling was done for cytogenetic analysis. The chromosomes were stained by Giemsa-trypsin banding and scanned using IKAROS software (MetaSystems, Altltussheim, Germany). The pedigree showed an autosomal recessive inheritance pattern with one additional affected male in the family. Twenty-five plates, which were screened for the proband, revealed normal karyotypes. However, genetic analysis could not be performed.
Case 2
A 41-year-old male presented with decreased vision, haloes, and visual distortion that he had been experiencing for the past 13 years. His brother (case 3) was known to have the same problems. The patient's best corrected visual acuity, with +0.50 DS / -4.50 DC ×100, was 6 / 36 in the right eye, and, with +0.50 DS / -4.00 DC ×80, was 6 / 15 in the left eye. Anterior segment examination was normal. Indirect ophthalmoscopy revealed vitreous floaters, macular schisis, and diffuse RPE alterations in both eyes. Also revealed was a peripheral hole in the inferotemporal quadrant in the left eye.
The patient's color vision was analyzed with the Farnsworth D-15 test [6]. The total error score for the Farnsworth test was 36 in the right eye and 19 in the left eye. The test results revealed tritanomaly in the right eye and diffuse color defect in the left eye. SD-OCT imaging of both eyes showed incomplete posterior vitreous detachment, elevated foveal contours, and foveal schisis. It also showed alteration of the photoreceptor layer. Foveal thickness was 292 microns in the right eye and 490 microns in the left eye. Microperimetry showed a reduced mean retinal sensitivity of 7.7 db in the right eye and 8.2 db in the left eye, with a central dense scotoma in both eyes (Fig. 2).
Case 3
A 36-year-old male presented with decreased vision, haloes, and visual distortion that he had been experiencing for the past 4 years. His brother (case 2) was known to have the same complaints. The patient's best corrected visual acuity, with +4.50 DS / -0.50 DC × 90, was 6 / 12 in the right eye, and, with +5.00 DS / -1.00 DC × 90, was 6 / 24 in the left eye. Anterior segment examination was normal. Indirect ophthalmoscopy revealed vitreous floaters, macular schisis, diffuse RPE alterations, and peripheral schisis in both eyes.
The patient's total error score for the Farnsworth D-15 color vision test was 28 in the right eye and 31 in the left eye. The test results revealed tritanomaly in the right eye and diffuse color defect in the left eye. SD-OCT imaging of both eyes showed epiretinal membrane. The foveal contour was altered with cystic spaces that were suggestive of retinal schisis. Alteration of the photoreceptor layer was noted. Foveal thickness was 136 microns in the right eye and 171 microns in the left eye. Microperimetry showed a reduced mean retinal sensitivity of 1.0 db in the right eye and 0.2 db in the left eye, with a central dense scotoma in both eyes (Fig. 3).
Discussion
Goldmann-Favre syndrome is characterized by foveal and/or peripheral retinoschisis. Differential diagnosis includes conditions such as retinitis pigmentosa (RP), Stickler's syndrome, and familiar exudative vitreo-retinopathy. RP is differentiated from Goldmann-Favre syndrome on the basis of vascular attenuation and electroretinogram. Cystoid macular oedema associated with RP is a potentially treatable condition, and can be distinguished from retinoschisis by OCT and FFA. Ocular management of Goldmann-Favre syndrome includes scatter photocoagulation of peripheral avascular lesions, as well as vitrectomy for clearance of vitreous hemorrhage and relief of retinal traction if present. Genetic counseling is also offered to patients and potential carriers. Because the syndrome is associated with mutations in Arg311Gln NR2E3 in the 15q23 chromosome, it is a potential target for gene therapy [7].
Case 1 followed the temporal sequence of events over a period of 18 months in a patient diagnosed with Goldmann-Favre syndrome. The patients in cases 2 and 3 were clinically diagnosed with Goldmann-Favre syndrome. We are unaware of previous reports about Goldmann-Favre syndrome that use SD-OCT and microperimetry for analysis. The findings vary with the age of the patient and the severity of the disease. Findings from SD-OCT imaging included lamellar macular holes, macular schisis, microcystic spaces, alterations of photoreceptor layers, epiretinal membrane with vitreomacular traction, enhanced foveal thickness, and elevated foveal contours. Microperimetry revealed reduced foveal sensitivity with dense scotomas. In this series of cases, retinoschisis and macular cystoid changes noted with SD-OCT matched the scotomas shown with microperimetry. Advanced diagnostic techniques such as SD-OCT and microperimetry detect new characteristics, as well as confirm correlations among known features, to elevate our knowledge of the disease.

 XML Download
XML Download