

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Trabecular meshwork cells play an important role in the regulation of aqueous outflow and open-angle glaucoma. Open angle glaucoma is known to occur with increased resistance of the outflow pathway due to the decreased cellularity and altered ultrastructure of the trabecular meshwork [1,2]. Free radical nitric oxide (NO) plays important physiological roles [3-5] and is involved in the regulation of outflow through the trabecular meshwork as the activation of NO synthetase lessens in glaucoma [6-9]. Decreased NO leads to the production of other intermediate species and covalently modified biomolecules that cause injury and cellular dysfunction. Down-regulation of endothelial nitric oxide synthase and increased generation of oxygen-derived free radicals leads to early inactivation of NO, which in turn promotes further generation of oxidants, possibly by uncoupling NO synthase. This vicious cycle provides an integrative explanation for the interrelation between NO deficiencies and an excess of oxygen-derived free radicals. The resultant increased oxidative stress is known to promote cellular senescence [10-12].
Advanced glycation end products (AGE) are formed by the non-enzymatic reactions of sugar and protein when the glucose concentration is abnormally elevated. AGE permanently accumulates in the blood vessels and then induces abnormal vasodilatation by restricting the physiological activation of NO in old age or in patients with diabetes mellitus [13,14]. Additionally, AGE are prevalent in ocular diseases such as cataracts or proliferative diabetic retinopathy [15,16]. Increased levels of AGE in the corneal endothelium of diabetes patients and in the aqueous humor in pseudoexfoliation glaucoma [17,18] suggests that AGE can accumulate in the trabecular meshwork and compromise its function. Davies et al. [19] reported that glucose levels in the aqueous humor of patients with diabetes were significantly higher (3.2 mM vs. 7.8 mM) compared with levels in persons without diabetes. Although diabetes and metabolic abnormalities are not known to be significant risk factors for glaucoma [20], the possibility of resultant oxidative stress causing cellular damage by AGE still exists since oxidative stress can occur as result of AGE accumulation [21].
The trabecular meshwork not only works as the outflow pathway, but also actively participates in the regulation of outflow. Since NO synthetase is expressed in the trabecular meshwork [22,23] and glucocorticoid receptors are found in trabecular meshwork cells [24], AGE could act on the trabecular meshwork and decrease NO generation.
If the NO generation is decreased by AGE, then intraocular pressure could be increased by inhibiting relaxation of the trabecular meshwork. In addition, if AGE generates oxidative stress and promotes the senescence of trabecular meshwork cells, then AGE may cause dysfunction of the trabecular meshwork. However, it is not yet known whether AGE has such an effect on human trabecular meshwork cells. Therefore, the effects of AGE on the generation of NO and oxidative stress, and on the senescence of trabecular meshwork cells were investigated in this study.
Materials and Methods
Cell culture
Tissues were extracted from the area surrounding the chamber angle of an eyeball obtained from the eye bank that had been extracted within six hours of death. The eye had not previously been afflicted with ocular disease nor had it undergone any type of surgery. The trabecular meshwork was stripped away from the anterior chamber angle and the explants were moved to a culture plate coated with poly-L-lysine (Sigma, St. Louis, MO, USA) and cultured in Dulbecco's modified Eagle's medium (DMEM; Gibco, Gaithersburg, MD, USA) containing antibiotics (Gibco) and 10% fetal bovine serum (Gibco) at 5% CO2. After confirming that the trabecular meshwork cells had grown out into the surroundings of the explanted tissues, the explants were removed and the culture was continued. When the cells were confluent, they were treated with trypsin and subcultured at a ratio of 1:3 with a medium containing 10% fetal bovine serum. The procedures performed were consistent with the tenets of the Declaration of Helsinki.
Preparation of advanced glycation end products-bovine serum albumin: glycated bovine serum albumin
Fetal bovine serum (50 mg/mL) without endotoxin, 100 mmol/mL glyceraldehyde and 1 mmol/L phenylmethylsulfonyl fluoride (Sigma) were dissolved in PBS solution. The mixtures were incubated under aseptic conditions for seven days after filtering. After dialyses with PBS solution, the electrolytes were removed by chromatography (Amersham PD-10 column chromatography; Amersham Pharmacia Biotech, Piscataway, NJ, USA). To determine the efficiency of glycation, browning of the glycated bovine serum albumin (G-BSA) preparation was determined by measuring the absorbance at 340 nm using a spectrophotometer. The absorbance of G-BSA increased with time, whereas the absorbance of control BSA (without glyceraldehydes) did not [25]. The concentration of G-BSA was measured with the Bradford assay and samples were stored in the deep freezer until use [26].
Drug treatment
After dividing the cells (1 × 105 cells/well of a 24-well culture plate, 0.6 × 105 cells/well of a 96-well culture plate), they were allowed to attach for 24 hours. The medium was removed and then replaced with DMEM medium including 1% fetal bovine serum and a low concentration of glucose (5 mM) to prevent the antioxidant effect of serum protein. The cells were exposed to 0, 10, 50, 100, and 200 µg/mL G-BSA for five days. A culture with 25 mM mannitol was simultaneously initiated to exclude the effects of osmotic pressure induced by AGE. The cells in the 24-well and 96-well plates were exposed to 100 µM L-arginine (Sigma) and 100 µM sepiapterin (Sigma), which are known to promote the NO generation. The cells were also exposed to an anti-oxidant of 50 µM N-acetylcysteine (NAC, Sigma) to reveal the effects of AGE on the generation of reactive oxygen species.
MTT and Griess assays
The effect of AGE on cell survival was assessed by 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyltetrazolium bromide (MTT, Sigma) assay [27], a colorimetric test frequently used as a screening tool to determine cell survival and toxicity, and NO generation was measured using the Griess assay [28]. For the MTT assay, the cultures were incubated for four hours after treatment with 100 µL of MTT per well, then washed with PBS solution and transferred to a 96-well culture plate. Then, 0.5 mL of dimethylsulfoxide was added to each well to dissolve the MTT formazan crystals. The absorbance was measured at 570 nm with a spectrophotometer (FLUOstar OPTIMA; BMG Labtech, Offenburg, Germany). Cell survival was computed as a percentage by dividing the absorbance values of the experimental group by the absorbance values of the control group, which was not treated. The Griess assay was performed to measure the quantity of nitrite, a reactant in NO generation. After mixing the medium containing cells with the same quantity of Griess reagent (Sigma), the absorbance was measured at 540 nm using a spectrophotometer. Serial dilutions of sodium nitrite (Sigma) were used to generate a standard curve.
Measurement of reactive oxygen species
The generation of superoxide was measured with a microplate reader using modified cytochrome c reduction [29,30]. The reactive solution was prepared by mixing 160 µM cytochrome c (Sigma) and 100 U/mL superoxide dismutase (Sigma), and cells were exposed to 100 µL of this solution for 20 minutes. The quantity of cytochrome c reduced at 540 nm was measured over the course of an hour. The quantity of superoxide was expressed as nmol/106cell/h, reflecting the quantity of cytochrome c (in nmol) reduced by 106 cells per hour with a 2.1 × 104/M/cm net extinction coefficient.
The generation of reactive oxygen species was measured using the dichlorofluorescin diacetate assay [31]. On the fifth day after treatment of cells in a 96-well culture plate, the medium was removed and cells were subsequently washed with PBS solution. Cells were then treated with 5 µM dichlorofluorescin diacetate (Sigma), after which they were incubated for 30 minutes at 37℃ in PBS in the dark. The change in fluorescence of oxidized dichlorofluorescin was measured at excitation and emission wavelengths of 488 nm and 527 nm, respectively, using a fluorescence analyzer (FLUOstar OPTIMA).
Assessment of apoptosis
To investigate whether AGE generates apoptosis, flow cytometry was performed by double staining of fluorescein isothiocyanite-labeled Annexin V/propidium iodide (PI) with a commercial kit (TACS Annexin V-FITC apoptosis detection kit; R&D systems, Minneapolis, MN, USA) [32]. After trypsinization and washing, 5 µL Annexin V and 1 µL PI were added to a 100 µL cell suspension and incubated at room temperature for 15 minutes, and then a 400 µL buffer solution was added and mixed. Next, flow cytometric analysis was performed (Cytomics 500FC; Beckman Coulter, Miami, FL, USA) at fluorescence emission wavelengths less than 530 nm and greater than 575 nm.
Assessment of cellular senescence
A commercial senescence-associated β-galactosidase kit (SA-β-gal, Sigma) was used to measure β-galactosidase activity, which is characteristic of senescent cells [33,34]. SA-β-gal is used to assess abnormal enzymatic activity observed in aging. The culture was incubated for two hours after adding staining solution; β-galactosidase in senescent cells stains blue. The proportion of senescent cells was computed as a percentage by dividing the number of senescent cells stained blue by the total number of cells. A round optical glass with a grid dividing the view into five square fields was attached to the eyepiece of an optical microscope and used to count the number of cells in each field over ten fields. The average numbers of stained and total cells were determined for these ten fields (×100).
Results
Cell culture
The trabecular meshwork cells started to grow out into the surroundings of the tissue explants as a monolayer after two or three weeks of primary culture. The presence of trabecular meshwork cells was confirmed by their characteristic morphology and their characteristic growth pattern as satellite colonies grew distant from the tissue explants [35,36].
Effects of advanced glycation end products on survival and nitric oxide generation
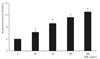
When trabecular meshwork cells were exposed to 100 µg/mL G-BSA for five days, cell survival was significantly decreased to 98.5% compared with non-exposed controls (p < 0.05) (Fig. 1). The decreased survival rate induced by G-BSA exposure was abolished by L-arginine and sepiapterin, which increase the generation of NO and NAC as antioxidants, respectively (p > 0.05) (Fig. 2). Mannitol appeared not to have a significant effect on cell survival and thus it was inferred that the effect of AGE was not due to a change in osmotic pressure (data not shown).
AGE significantly decreased the quantity of NO generated in the medium (p < 0.05) (Fig. 3). The decrease in NO generation was abolished by L-arginine, sepiapterin and antioxidants (p > 0.05) (Fig. 4). Thus, the decrease in the survival of trabecular meshwork cells caused by AGE could be caused by a decrease in NO or induced oxidative stress.
Effects of advanced glycation end products on the generation of reactive oxygen species
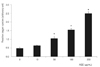
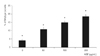
Treatment with 10 µg/mL AGE significantly increased the generation of superoxide in trabecular meshwork cells (p < 0.05) (Fig. 5). The increased generation of superoxide was abolished by L-arginine, sepiapterin and NAC (p < 0.05) (Fig. 6). AGE increased the generation of reactive oxygen species (p < 0.05) (Fig. 7), and this increase in reactive oxygen species was abolished by NAC (p < 0.05) but not by L-arginine or sepiapterin (p > 0.05) (Fig. 8).
Effect of advanced glycation end products on the cell apoptosis and senescence
On FITC-Annexin/PI double staining, normal live cells were FITC-negative and PI-positive, early apoptotic cells were FITC-positive and PI-negative, and necrotic cells were FITC-positive and PI-positive. Flow cytometric analysis revealed that apoptosis had been induced in cells exposed to 10 mg/ml G-BSA for 5 days (p < 0.05) (Fig. 9). The apoptotic cell population was 7.57 ± 0.5% in the control group (without AGE treatment). In the experimental group, there was a dose-dependent increase in the apoptotic cell population in the following order: 50 µg/mL (11.39 ± 2.4%), 100 µg/mL (16.61 ± 1.0%), and 200 µg/mL (26.91 ± 2.0%).
SA-β-gal staining was performed to investigate the effect of G-BSA on cell senescence. Senescent cells stained blue and began to appear after exposure to 10 µg/mL G-BSA (Fig. 10). As the concentration of G-BSA increased, senescent cells were observed more frequently (Fig. 11). When cells were co-exposed to L-arginine, sepiapterin and NAC with 50 µg/mL and 100 µg/mL G-BSA, the number of senescent cells significantly decreased compared to the cells exposed to G-BSA only (Fig. 12). Thus, it appears that AGE induces apoptosis and promotes the senescence of trabecular meshwork cells.
Discussion
This study shows that AGE not only decreases cell survival by inducing apoptosis, but also promotes the senescence of trabecular meshwork cells accompanied by increased oxidative stress. AGE is a composite substance formed by non-enzymatic glycation that changes the structure and the function of proteins. The permanent glycation of protein is closely linked to oxidative stress. AGE is known to increase the generation of reactive free radicals by a process similar to lipid peroxidation, generate oxidative stress in cells by activating AGE membrane receptors, and induce aging, glycosuria, vascular disease and nervous atrophy by promoting the inflammatory response [37,38]. AGE is also related to various ocular diseases including diabetic retinopathy [39-43] and it is known to accumulate in the optic nerve head [44].
Dysfunction of the trabecular meshwork increases the resistance of the outflow pathway and induces an increase in intraocular pressure. It is important to maintain the normal physiological function of the trabecular meshwork because it functions in regulating outflow [20]. The number of trabecular meshwork cells decreases with aging [45] and the histopathologic findings of primary open-angle glaucoma tissue are similar to those of aged cells [46-49].
Although NO acts as an important physiological adjusting factor in the human body at low concentrations, it is converted into reactive oxygen species and may induce pathologic damage at either excess or deficient states [3-5]. The decreased efficiency of NO in the trabecular meshwork not only decreases outflow as the trabecular meshwork shrinks, but can also cause various pathologic consequences as a result of decreasing physiological activation. When the physiological activation of NO decreases, the generation of harmful reactive oxygen species such as superoxide or peroxynitrite increases [50]. These reactive oxygen species not only induce oxidative stress but also bring about the senescence of cells [51-56]. In addition, ascorbic acid, which exists in high concentrations within the aqueous humor, plays a role as an antioxidant by decreasing oxidative stress and, thereby decreasing cellular senescence [57].
In this study, G-BSA was used as an AGE because glycolaldehyde has previously been reported as the best modifier for fast and reproducible generation of a high-affinity receptor for AGE binding and AGE [25]. Our results show that AGE decreased the production of NO in trabecular meshwork cells. Thus, trabecular outflow may be decreased by contraction or inhibition of the relaxation of the trabecular meshwork. AGE also increased the generation of superoxide and reactive oxygen species. Superoxide could possibly be further increased by the uncoupling of NO, and promoting the generation of harmful free radicals such as peroxynitrite may cause further damage [58]. Thus, if the generation of NO is promoted at the physiological level or the synthesis of reactive oxygen species is restrained, it may be helpful to keep the trabecular meshwork functionally intact to prevent further damage. As the results of this experiment demonstrate, L-arginine and sepiapterin, which participate in the generation of NO or NAC as antioxidants, not only increased the generation of NO, but also decreased the generation of reactive oxygen species. These results indicate that NO plays a role in keeping the outflow pathway open by relaxing the trabecular meshwork and in preserving the functionality of the trabecular meshwork, which is continually exposed to free radicals in the aqueous humor. The role of NO and the importance of oxidative stress as the mechanism underlying glaucoma have been presented in recent studies [59,60].
Although AGE-induced generation of superoxide is suppressed by either NO supplements or antioxidants, AGE-induced generation of reactive oxygen species is suppressed by antioxidants only. AGEs accelerate the formation of superoxide, which generates peroxynitrite by reacting with NO. The availability of NO is related to superoxide, one of the mitochondrial reactive oxygen species. Therefore, the increased availability of NO by supplementation of L-arginine and sepiapterin with inhibition by the uncoupling of eNOS could result in the reduced generation of superoxide. Our results are consistent with previous studies that found that AGE enhanced peroxynitrite formation mediated by NO and superoxide in retinal neurons [58,61,62]. Various reactive oxygen species other than superoxide are formed by AGE and this could lead to oxidative stress unrelated to NO or superoxide. Our findings indicate that this AGE-induced oxidative stress could be reduced by antioxidants.
Reactive oxygen species not only damage trabecular meshwork cells, but also induce apoptosis and promote cellular aging, and NO is known to play a role in delaying cellular senescence [10-12]. Trabecular meshwork cells of the human eye have been suggested as the proper model system for the study of the cellular ageing phenomenon [63]. It was reported that the number of trabecular meshwork cells decreases due to tissue damage induced by oxidative stress [64-67] and senescence in glaucoma [68,69]. In this study, protein glycation appeared to induce functional changes in cells and AGE appeared to promote the senescence of trabecular meshwork cells. The exacerbation of trabecular meshwork cell senescence by AGE could be caused by oxidative stress supported by NO generation and the restraint of cell senescence by antioxidants.
To conclude, AGE induced the senescence of cultured human trabecular meshwork cell, mediated by an increase in oxidative stress. Thus, maintaining NO generation at the physiological level or using antioxidants makes the trabecular meshwork competent not only by maintaining the patency of the trabecular meshwork as an outflow pathway, but also by decreasing damage to and senescence of the trabecular meshwork due to oxidative stress. The senescence of trabecular meshwork cells due to oxidative stress could serve as a target for a therapeutic strategy to preventing damage in the management of glaucoma.


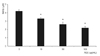






 XML Download
XML Download