

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Monoclonal gammopathy of undetermined significance (MGUS) is a benign asymptomatic plasma cell neoplasm containing monoclonal protein (M protein). MGUS is defined as a serum M protein level less than 3.0 g/dL and bone marrow plasma cell level less than 10% [1]. Diagnosis of this disease is made by detection of M protein in blood or urine samples. Since MGUS is asymptomatic, it is usually detected incidentally during routine laboratory evaluation [2].
Corneal involvement in monoclonal gammopathy is a very rare complication and is difficult to diagnose. According to Garibaldi, corneal disease was detected in one of 100 monoclonal gammopathy patients [3]. Corneal opacities occurring in various layers caused by abnormal protein deposition are the most common features, and these deposits can appear in many shapes when examined under electron microscopy. In most cases, there are no symptoms or minimal foreign body sensations and glare. Few cases of MGUS patients have been reported to show corneal crystalline deposits. We report here on a severe case of crystalline keratopathy caused by monoclonal gammopathy in a patient who underwent three penetrating keratoplasty procedures.
Case Report
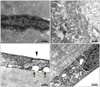
A 62-year-old female presented to our clinic in July 1996 with progressively decreased visual acuity and pain in both eyes that had begun 12 years prior. Her visual acuity was counting fingers at a distance of 50 cm from the right eye, while that of the left eye was 0.06. Corneal stromal edema and Descemet's folds were found in both eyes (Fig. 1A). Corneal opacity was involved in all layers of the cornea, and epithelial bullas were found in the center of the right cornea. Lens, vitreous, and fundus were normal. The patient had neither a personal medical history nor familial history of eye disease. We performed penetrating keratoplasty in the right eye without a precise diagnosis. Visual acuity was maintained at 0.2 during the first few months after surgery.
Visual acuity proceeded to decrease, and ocular pain recurred four years after the first surgery. Right eye visual acuity was hand motion, and slit-lamp biomicroscopy showed recurrence of epithelial edema and stromal opacity (Fig. 1B). In July 2005, the patient underwent a second penetrating keratoplasty in the right eye due to recurrent corneal opacity and neovascularization in the cornea. Despite mild corneal opacity and corneal edema, visual acuity was maintained at 0.08 during the first two years after the second surgery.
A third penetrating keratoplasty was performed on the right eye in July 2008, due to worsened corneal opacity and visual acuity decrease to 0.02 (Fig. 1C). Total serum protein level, which had remained at a normal level until 2008, was elevated to 10.4 g/dL after preoperative evaluation. Electrophoresis revealed elevated M protein in the serum (2.72 g/dL) and urine (29.3 mg/dL). Immunofixation electrophoresis showed elevated kappa light chains. Bone marrow smears depicted a plasma cell concentration of 2.4%, and there were no morphological abnormalities. The patient was diagnosed with type IgG-kappa MGUS and was transferred to the hemato-oncology department for further examination.
In the hemato-oncology department, corneal biopsy of the right eye was performed. The corneal epithelial layer showed loss of polarity and clusters of epithelial cells (Fig. 2A). A decreased number of endothelial cells accompanied by the presence of vacuoles in the Descemet's membrane were detected (Fig. 2B). TUNEL staining typically shows condensed or fragmented nuclei in apoptotic cells. In this case, TUNEL staining of the cornea showed increased keratocyte apoptosis with condensed nuclei (Fig. 2C). Numerous eosinophilic deposits were stained bright red with Masson trichrome stain and were thought to be IgG-kappa light chains, the levels of which were elevated in this patient (Fig. 2D).
Electron microscopy depicted numerous microtubular-shaped deposits located extracellulary near corneal epithelial cells, stromal cells, and Descemet's membrane. These microtubular deposits surrounded keratocytes, which contained condensed chromatin (Fig. 3A). A portion of these deposits appeared to be phagocytosed by macrophages (Fig. 3B). However, in monoclonal gammopathy, phagocytosis is not sufficient to remove all abnormal crystal deposits. In addition, vacuoles and loss of microvilli in endothelial cells and abnormal banded collagen fibrils in the posterior portion of Descemet's membrane were observed (Fig. 3C and 3D). This demonstrates that deposition of abnormal protein crystals can affect not only keratocytes but also endothelial cells and the collagen matrix in the stroma.
Discussion
Monoclonal gammopathy-associated crystalline keratopathy was first reported in a multiple myeloma patient by Meesman in 1934 [4]. Only 1% of monoclonal gammopathy patients show crystal deposits, including a case of a multiple myeloma in a patient from Korea in 2009 [5]. Primary symptoms include corneal crystal deposits and corneal opacity. Corneal opacity may be the first clinical symptom of monoclonal gammopathy and affects the corneal epithelium and stroma. Endothelial dysfunction plays a major role in corneal opacity, but the association between corneal endothelial dysfunction and crystal deposition is not clear. Although there have been some reports of decreased corneal opacity with systemic treatment, some patients still require penetrating keratoplasty.
Extracellular deposition of M protein creates the crystals found in the cornea. These crystal depositions can involve all layers of the cornea and vary in shape (e.g., rectangular or hexagonal), location (corneal epithelium, stroma, endothelium, conjunctiva and lens), and resulting symptoms [4]. The typical shape of crystals observed under electron microscopy is the 10 nm 13 nm parallel band shape [6-8]. The etiology of this crystal deposition is not clear. However, from the pathological findings of this case, we can assume that phagocytic activity in the cornea is insufficient to manage the monoclonal proteins so that these over-produced proteins are deposited in extracellular areas and lead to keratocyte apoptosis.
In this case, diagnosis was complicated due to differences in clinical presentation after each penetrating keratoplasty. The diagnosis of crystalline keratopathy was made after MGUS was confirmed and crystalline deposits had been detected. Because corneal crystal deposition is a rare finding in monoclonal gammopathy patients, various diagnoses such as lattice dystrophy, Schnyders crystalline dystrophy, granular dystrophy, and cystein deposition can be suspected, particularly when crystal deposition presents as the first symptom [9-12].
It is clinically difficult to distinguish crystalline keratopathy from the other diseases above. Histological examinations of corneal biopsy and systemic evaluations to confirm the crystal-producing condition are required to make a firm diagnosis. This is a good example of an ophthalmologic case that requires systemic evaluation in surveillance of systemic disease progression or for measurement of disease severity.
Corneal allograft rejection and recurrence of corneal crystal deposition can be distinguished easily in the clinic. The main difference between rejection and recurrence is the rate of progression. Corneal graft rejection most commonly occurs during the first year after surgery and presents with the severe and acute symptoms of blurring of vision, eye redness and pain. In contrast, corneal crystal depositions occur relatively slowly and cause less severe symptoms, and most corneal crystal depositions are asymptomatic. However, some cases can cause progressive pain and blurred vision. As in this case, fast progressing and painful corneal crystal depositions can only be distinguished from corneal allograft rejection via corneal biopsy.
The current standard of care for MGUS is observation without treatment. Patients with MGUS need to be rechecked after 3-4 months for progression of MGUS to multiple myeloma, amyloidosis, or non-Hodgkin's lymphoma. According to Kyle [13], the progression rate of MGUS to malignant disease is 1% per year.
Although MGUS is a benign plasma cell disorder, crystalline keratopathy can occur and may progress to severe vision loss. Because MGUS is an asymptomatic disease, and only decreased vision with corneal opacities can be found in the early presentations, a systemic work-up that includes serologic tests such as serum protein or cholesterol level is needed for patients with unexplainable corneal opacity. On the other hand, the eyes of patients diagnosed with monoclonal gammopathy should be carefully examined. Adequate treatments, including surgery, are often required.


 XML Download
XML Download