

 PDF
PDF ePub
ePub Citation
Citation Print
Print


The triad of spinocerebellar ataxia, chorioretinal dystrophy, and hypogonadotropic hypogonadism (Boucher-Neuhäuser syndrome) has been described as an autosomally inherited recessive disorder.1 Only five families have been reported to date,1-5 but none of them in the ophthalmic literature. We studied the ophthalmologic findings before the neurologic symptoms in this additional patient with this rare disorder.
Case Report
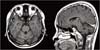
An 18-year-old Asian man had experienced decreased vision in both eyes over the past 6 years. His medical history indicated delayed puberty. At 16 years of age, the patientexperienced progressive deterioration of his balance and gait disturbance. Magnetic resonance imaging of the brain showed diffuse atrophy of the cerebellar hemispheres (Fig. 1).
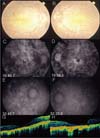
At 12 years of age, he first noticed progressive loss of vision and photophobia in both eyes. The best-corrected visual acuity was 20/200 bilaterally. There was no blepharoptosis, and the extraocular movements were full with vertical and horizontal gaze-evoked nystagmus. The anterior segment examination was normal. Ophthalmoscopy showed extensive atrophic changes of the retinal pigment epithelium and choriocapillaris in the posterior pole and midperiphery of both eyes, with visible choroidal vessels in some areas. Some clumps of pigment deposition (without bone spicule configuration) were also seen. The retinal vessels and optic disc appeared normal (Fig. 2A, B). Fluorescein angiography showed hypofluorescence with large, visible choroidal vessels in areas showing retinal pigment epithelial and choriocapillaris atrophy (Fig. 2C, D).
Indocyanine green angiography showed normal fluorescence of the retinal arteries and ground-glass fluorescence of the choriocapillaris (Fig. 2E, F). An Ishihara test identified considerable abnormality in color discrimination. Perimetry demonstrated a ring scotoma at 10 to 60° from the fixation points in both eyes.
Optical coherence tomography showed retinal thinning, blunting of the foveal pit, and disappearance of the outer nuclear layer. The foveal center measured 204 µm in the right eye and 174 µm in the left (Fig. 2G, H).
An electroretinographic examination showed subnormal photopic and scotopic responses bilaterally (Fig. 3). The photopic responses to both high-intensity white flash and 30-Hz flicker stimuli showed 20 and 40% reductions from the mean b-wave amplitude, respectively. The dark-adapted scotopic responses to high- and low-intensity flash stimuli showed prolonged implicit times. Electroretinographic oscillatory potentials were absent, reflecting a reduction in the vascular supply to the retina.
On general examination, the patient was 176 cm tall and weighed 64 kg. He had normal male secondary sex characteristics. Hormone studies were consistent with hypogonadotropic hypogonadism. His intelligence was normal. His gait was wide-based and ataxic. Romberg's sign was absent. Laboratory findings, including plasma and urine amino acids, phytanic acid, and lipid plasma levels, were all normal. There was no family history.
Discussion
The clinical findings in this patient are consistent with Boucher-Neuhäuser syndrome. The absence of manifestations in the patient's siblings suggests that this disorder is inherited in an autosomal recessive pattern, as previously reported.2 This case showed the main ocular manifestations mentioned in reported cases, including atrophic changes of the retinal pigment epithelium and choriocapillaris, with clumps of pigment deposition in the posterior pole and midperiphery. Electroretinographic changes, with a reduction in the amplitudes of both the rod and cone responses have also been found in all reported cases.
Baronicini et al.4 described what appeared to be the earliest ophthalmoscopic and electroretinographic changes in the asymptomatic 6-year-old brother of their propositus, who subsequently showed the classic chorioretinal findings at the age of 30 years. These early changes consisted of fine pigmentary alterations throughout the retina, with predominant involvement of the posterior pole and coarse pigmentation of the macula. Although his visual acuity was 20/20 in both eyes, electroretinography at that time already showed subnormal photopic and scotopic responses.
In this case, the visual complaints appeared before thehypogonadism and spinocerebellar ataxia. The first areas of chorioretinal degeneration appeared at the age of 12 years. Ocular involvement may be the first manifestation of this triad, as the onset of visual complaints in reported cases ranges from the first to sixth decade of life.4 Similarly, the expression and progression of the chorioretinal degeneration varies widely, as does the visual acuity in reported cases, ranging from 20/30 to 20/200.4,5 This case exhibited abnormal color discrimination. However, others have reported essentially normal color discrimination in a patient with advanced chorioretinal lesions.5 Additional reported ocular manifestations, which were present in this patient, include nyctalopia and nystagmus in the primary position of gaze.4,5 Although no ocular histopathological studies of this disorder have been reported, the clinical and electrophysiological data suggest that the degeneration involves primarily the outer retinal layers, retinal pigment epithelium, and choriocapillaris, together with alterations of the vascular supply to the retina.
The neurologic signs usually develop as slowly progressive or non-progressive ataxia in the third decade of life.4 Other disorders in which cerebellar ataxia is associated with ocular fundus changes in adults include olivopontocerebellar atrophy type III, Refsum's disease, and abetalipoproteinemia. However, olivopontocerebellar atrophy type III is an autosomal dominant disorder, and the degeneration involves only the external layers of the retina and the retinal pigment epithelium, and not the choriocapillaris.6 In addition, diffuse changes of the fundus usually appear only in infantile-onset disease, and severe neurologic symptoms and early death are common in these patients.7,8 In late-onset olivopontocerebellar atrophy type III, usually only the macula is affected.6,8 In this case, we excluded this disorder because of the extensive involvement of the choriocapillaris and the absence of a family history of visual or neurologic abnormalities. Furthermore, he showed hypogonadism, a finding not associated with olivopontocerebellar atrophy.
The clinical features of Refsum's disease include cerebellar ataxia and retinal dystrophy with pigment changes, bone-spicule formation, optic atrophy, and attenuated retinal vessels.9 Normal phytanic acid levels in this patient and the lack of other common findings in Refsum's disease, such as ichthyosis, nerve deafness, anosmia, and epiphyseal dysplasia, led us to exclude that diagnosis.
Abetalipoproteinemia (Bassen-Kornzweïg syndrome) is also associated with ataxia and retinal dystrophy.10 However, other common findings, such as acanthocytosis, malabsorption syndrome, cardiac abnormalities, and low lipid plasma levels, were absent in this case.
When the triad of hypogonadotropic hypogonadism, spinocerebellar ataxia, and chorioretinal degeneration is present, and other siblings are affected, the diagnosis of Boucher-Neuhäuser syndrome is relatively easy. However, the diagnosis may be difficult to establish when visual complaints occur in a prepubertal child or when they appear before the neurologic symptoms.11 The Boucher-Neuhäuser syndrome should be included in the differential diagnosis of patients with chorioretinal degeneration, particularly if there are neurologic or endocrinologic manifestations.

 XML Download
XML Download