

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Weill-Marchesani syndrome (WMS) is a rare connective tissue disorder first described by Weill1 in 1932, and further delineated by Marchesani.2 Alternatively it has been named spherophakia-brachymorphia syndrome, or congenital mesodermal dysmorphodystrophy. Diagnositc criteria of WMS includes: (1) short stature; (2) brachydactyly; (3) microspherophakia and/or ectopia lentis.3 These patients may have joint stiffness and heart defects. Most patients have been described by ophthalmologists, since ocular symptoms and signs are characteristic of this syndrome and call for clinical attention. Characteristic eye abnormalities consist of dislocation of the microspherophakic lens, which causes high myopia, acute and/or chronic angle-closure glaucoma and cataracts. Despite the disease's clinical homogeneity, autosomal recessive and autosomal dominant modes of inheritance have been reported.4,5
Here, we report a sporadic case of WMS, presenting with corneal endothelial dysfunction due to a dislocated lens and corneolenticular contact. The spherophakic lens was morphologically confirmed by a rotating Scheimpflug camera and ultrasound biomicroscopy. Lensectomy with intraocular lens fixation was performed for visual rehabilitation.
Case Report
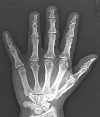
A 17-year-old woman was referred to our clinic with high myopia and progressive visual disturbance. Her height was 147 cm, and her body weight was 50.6 kg. She had brachydactyly and short metacarpal bones on X-ray images of the hand (Fig. 1). Initial Snellen uncorrected visual acuity (UCVA) was 2/200 in both eyes, and best corrected visual acuity (BCVA) was 20/50 (-sph 20.50 -cyl 3.00 Ax 180) in her right eye and 20/40 (-sph 16.00 -cyl 6.00 Ax 30) in her left eye. A-scan biometry revealed that the axial lengths of her eyeballs were 23.65/23.30 mm, suggesting a lenticular origin for her myopia. She had been treated with bilateral laser peripheral iridotomies at a local clinic two years ago. Despite this treatment, her initial intraocular pressure (IOP) was 26/22 mmHg by Goldmann applanation tonometry. Slit lamp examination of both eyes before pupillary dilation showed edematous corneas, extremely shallow anterior chambers, open peripheral iridectomy and iridocorneal/ corneolenticular contact. After pupillary dilation, IOP decreased to 22/19 mmHg, due to ciliary muscle relaxation. Slit lamp examination revealed bilateral superonasal subluxation of the crystalline lens with the lens equator and zonule visible within the pupil (Fig. 2). Central corneal edema was observed because the anteriorly dislocated lens was touching corneal endothelium. Due to corneal endothelial dysfunction, endothelial cell counts (743/675 cells per mm2) and hexagonality (33/0%) were decreased on specular microscopy (Cell Chek®, Konan, Tokyo, Japan) (Fig. 3), and central corneal thickness was increased to 672/712 µm on ultrasonic pachymeter (UP-1000®, Nidek, Maehama, Japan). On rotating Scheimpflug camera (Pentacam®, Oculus, Wetzlar, Germany) examination, increased anteroposterior diameter of the spherophakic lens (5200/5020 µm), hypoplastic ciliary body, elongated and stretched zonules, and the anteriorly dislocated lens touching central corneal endothelium were clearly demonstrated (Fig. 4). Ultrasound biomicroscopic assessment (HiScan®, Optikon, Rome, Italy) revealed a steep anterior lens curvature, angle narrowing, iridocorneal and corneolenticular contact, hypoplastic ciliary body and elongated zonules (Fig. 5). Chromosomal analysis of phytohemagglutinin stimulated peripheral lymphocytes by Giemsa banding technique revealed 46, XX, inv (15) (q13qter) (Fig. 6). The patient's parents and two sisters demonstrated normal cytogenetic examination and crystalline lens morphology.
Echocardiographic examination showed trivial mitral regurgitation and tricuspid regurgitation, but her left ventricle ejection fraction (LVEF: 65%) and gross cardiac morphology were normal. Systemic evaluation was done to exclude homocystinuria, Alport syndrome, mandibulofacial dysostosis, hyperlysinemia, sulfite oxidase deficiency, Marfan syndrome, and Kleinfelter syndrome.
From these findings, we diagnosed this patient with a sporadic case of WMS with inversion of chromosome 15, who also showed the corneal endothelial dysfunction due to corneolenticular contact.
Since this patient showed impending corneal decompensation, surgical extraction of the crystalline lens was performed in the left eye. After 3.2 mm-sized scleral tunnel incision, phacoemulsification and irrigation/aspiration of the nucleus and cortex were performed in the capsular bag. The wound was enlarged to 7.0 mm for sclera-sutured sulcus fixation of a polymethyl methacrylate intraocular lens (CZ70BD, Alcon, Fort Worth, USA). The intraocular lens was sutured to the sclera 1.0 mm from the limbus with a 10-0 polypropylene suture. Two weeks later, the same procedure was performed in the right eye. One month postoperatively, the visual acuity was 20/40 without correction in both eyes. Three months after surgery, UCVA was 20/30 (OD) and 20/40 (OS), and BCVA was 20/20 (+sph 0.50 -cyl 2.00 Ax 160 : OD), 20/25 (+sph 1.50 -cyl 3.00 Ax 30 : OS). On slit lamp examination, the cornea was clear and the anterior chamber had deepened (Fig. 7). Endothelial cell counts had increased to 1774 / 2882 cells per mm2 on specular microscopy. Six months postoperatively, the same visual acuities and endothelial cell counts were maintained (Table 1).
Discussion
WMS is a rare connective tissue disorder characterized by short stature, brachydactyly, and spherophakia. Its mechanism is thought to be due to a developmental abnormality of mesodermal origin tissues (ciliary body, lens, and epiphysis of bones). It is speculated that in spherophakia, the fetal lens which is physiologically spherical, had never been subjected to the force of a properly acting ciliary body and zonules.6
Two authors have reported isolated spherophakia cases in Korea.7,8 In those cases, the cornea was clear and there were no other systemic abnormalities. Their main problems were visual disturbances due to lenticular myopia and IOP elevation. Those patients received bilateral extracapsular lens extraction with/without trabeculectomy due to IOP elevation, intraocular lenses were not implanted, and visual improvement was not remarkable postoperatively. In contrast with previous reports, we presented a typical WMS case with genetic abnormalities and characteristic external appearances, including short stature and brachydactyly. Our case showed that the zonules were extremely elongated enough to produce impending corneal decompensation due to corneolenticular contact. In addition, we achieved successful visual rehabilitation and prevented threatening visual conditions by prompt surgical interventions. We also demonstrated a hypoplastic ciliary body, elongated zonules, and a spherophakic crystalline lens with the aid of modern imaging devices such as ultrasound biomicroscopy and rotating Scheimpflug camera.
Despite the disease's clinical homogeneity, autosomal recessive (AR) and autosomal dominant (AD) modes of inheritance have been reported.4,5 In a literature review of 128 cases of WMS-Including 57 AR cases, 50 AD cases, and 21 sporadic cases-Faivre et al.3 found no significantdifferences in the mode of inheritance for patients with short stature, brachydactyly, mental retardation, myopia, and glaucoma. Some differences were found for microspherophakia (94% in AR, 74% in AD), ectopia lentis (64% in AR, 84% in AD), joint limitation (49% in AR, 77% in AD), and cardiac anomalies (39% in AR, 13% in AD). Some heterozygotes for the AR form presented with mild clinical manifestations of the disease.9 These results underscore the difficulties of genetic counseling in sporadic cases.
Genetic mutations in WMS have been mapped to FBN1 (fibrillin-1, gene map locus 15q21.1),10 ADAMTS-10 (A disintegrin-like and metalloproteinase with thrombospondin type 1 motif 10, gene map locus 19p13.3-p13.2),4 and ADAMTS-1 (gene map locus 21q21.2).11
ADAMTS metalloproteinase is involved in various human biological processes (normal or pathological), including connective tissue organization/turnover, coagulation, inflammation, arthritis, angiogenesis, and cell migration.12 It has recently gained attention with the discovery of its role in a variety of diseases, including tissue and blood disorders, cancer, osteoarthritis, Alzheimer's and the genetic syndrome autosomal recessive WMS (ADAMTS-10), thrombotic thrombocytopenic purpura (ADAMTS-13), and Ehlers-Danlos syndrome type VIIC (ADAMTS-2).13
Fibrillin, encoded by a locus on human chromosome 15, is the major constitutive element of extracellular microfibrils and has widespread distribution in both elastic and nonelastic connective tissue throughout the body.14 Fibrillin was immunolocalized to the ciliary zonule, a ligamentous structure consisting mainly of fibrillin microfibrils that is characteristically affected in Marfan syndrome and WMS.15 Mutations in the FBN1 gene are the major cause of Marfan syndrome and autosomal dominant WMS.10 In this case, there was an inversion of chromosome 15, which might be associated with the fibrillin abnormality. To confirm this genetic mutation, allele-specific RT-PCR reaction analyses or direct sequencing of genome are required because the fibrillin gene is large and the coding sequence is highly fragmented (65 exons).10
Angle-closure glaucoma can occur in spherophakia through the pupillary block mechanism caused by the dislocation of the lens into the anterior chamber or forward movement of the lens, which depends on the zonular integrity.16 When zonules are intact, the anterior lens curvature and forward positioning of the lens can lead to iridolenticular contact and pupillary block. The zonules are typically long in WMS, and the loosening of the zonules allows the globular lens to move forward, producing corneolenticular contact and pupillary block. Glaucoma is mainly reported in the literature mainly when spherophakia is associated with WMS.17 Glaucoma in isolated and familial spherophakia is less widely reported.18
In this case, the initial IOP (26/22 mmHg) were decreased to 22/19 mmHg by pupillary dilation. Pupillary block is exacerbated with miotics and relieved by mydriatics. Urbanek19 introduced the term inverse glaucoma to describe this phenomenon. Miotics cause ciliary muscle contraction and loosening zonular support, allowing forward movement of the lens and shallowing of the anterior chamber, eventually leading to pupillary block. Cycloplegics relax the ciliary mucle, tighten zonular support, and cause posterior lens movement. If miotics do not cause an IOP rise, the lens is probably dislocated with poor zonular support.17 Laser peripheral iridotomy can relieve pupillary block, and the area of appositional angle closure can be opened. If pupillary block in spherophakia remains undiagnosed, and if treatment (e.g. iridotomy) is delayed, permanent peripheral anterior synechiae and irreversible damage to the trabecular meshwork may result. In this circumstance, the IOP may remain elevated after iridotomy, and can be difficult to control.17
In this case, due to impending corneal decompensation by corneolenticular contact and chronic IOP elevation despite peripheral iridotomy, there were no alternatives other than prompt surgical intervention. By surgical lensectomy and intraocular lens implantation, decreased corneal thickness, increased corneal clarity, increased corneal endothelial counts, and good IOP control could be achieved. Increased endothelial counts and hexagonality suggest that peripheral endothelial cells had migrated and redistributed because of the elimination of corneolenticular contact. This case shows that even in complicated cases and under suboptimal conditions, it is possible to achieve good visual rehabilitation with rapid intervention and modern surgical techniques.
Modern imaging devices can be helpful in easily diagnosing this rare disease. By using slit lamp examination only, we can make a diagnosis of spherophakia roughly. However, it is hard to evaluate the zonular integrity and exact status of corneolenticular contact. As mentioned earlier in this article, the fetal lens, which is physiologically spherical, has never been subjected to the force of a properly acting ciliary body and zonules.6 The pathology of spherophakia is related mainly to the weakened zonule due to fibrillin abnormalities. Moreover, it is important to assess the zonular integrity to decide the proper surgical treatment plan. There have been reports of spherophakia assessment using ultrasound biomicroscopy.6,18,20 In this case, we used ultrasound biomicroscopy and a rotating Scheimpflug camera to confirm WMS. This is the first report of WMS assessed by the rotating Scheimpflug camera. Rotating Scheimpflug camera provided higher definition and clearer images of whole anterior segment structures. The whole shape of the microspherophakic lens touching corneal endothelium, hypoplastic ciliary body, and elongated zonules could be clearly visualized with the rotating Scheimpflug camera. For an examination using ultrasound biomicroscopy, the immersion probe should be in contact with cornea, and the patient should be in prone position. So it is difficult to evaluate the accurate status of corneolenticular contact, because the immersion probe may compress the cornea, and the dislocated spherophakic lens may be posteriorly displaced in supine position. A rotating Scheimpflug camera can be measured without corneal contact, with the patient in the sitting position, and requires less time overall. Interestingly, by comparing two imaging devices, we found that the rotating Scheimpflug camera showed bilateral corneoleticular contact in sitting position (Fig. 4), but in ultrasound biomicroscopy, the left eye showed corneolenticular contact, and the right eye showed only iridocorneal contact without corneolenticular touch, by the posterior shifting mechanism of the lens in supine position (Fig. 5). This result means that continuous corneolenticular contact was present in her left eye, while only intermittent contact occurred in her right eye. In her left eye, the corneal endothelial count was less(743/675 cells/mm2) and the central corneal thickness was thicker (672/712 µm) than those of her right eye. For that reason we performed the operation on the left eye first. Consequently, utilizing these modern imaging devices can be helpful in accurately diagnosing the morphologic status of WMS, which cannot be identified on slit lamp examination only and also in making a decision regarding the optimal surgical intervention method and timing.
In conclusion, WMS with chromosomal abnormality may cause severe corneal endothelial dysfunction due to a dislocated spherophakic lens. Prompt lensectomy is thus important in preventing such complications.






 XML Download
XML Download