

 PDF
PDF ePub
ePub Citation
Citation Print
Print


Experimental autoimmune uveoretinitis (EAU) is a prototypic T-cell-mediated autoimmune disease in which the target tissue is the neural retina. EAU is characterized by granuloma formation in the neural retina, destruction of photoreceptor cells, and blindness.1,2 The pathology of EAU in an animal model of putative autoimmune etiology serves as a model for sight-threatening posterior uveitis or panuveitis such as Vogt-Koyanagi-Harada (VKH) disease, Behcet's disease, birdshot retinochoroidopathy, sympathetic ophthalmia, and ocular sarcoidosis.3 There was a report that oxidative stress induced HO-1 exclusively in the Muller cells of mouse retina in vitro study,4 but others report that HO-1 expressed in the rat retina against light damage.5 Ohta et al.6 reported that there was a protective role of HO-1 against endotoxin-induced uveitis (EIU). They revealed HO-1 and HO-2 was expressed in iris and ciliary body (ICB). EIU is an animal model corresponds to anterior uveitis that is improved easily by topical steroid therapy. However, this study is first time to disclose that HO-1 has an important role against EAU that correspond to posterior uveitis.
Oxidative stress is the fundamental mechanism underlying many different diseases, and is implicated in the pathogenesis of many neurological pathologies. Organisms use a multitude of endogenous antioxidant mechanisms to defend against the deleterious effects of oxidative stress. Heme oxygenase-1 (HO-1) is one such potent mechanism. HO-1 protein, also called heat shock protein 32 (hsp32), is the rate-limiting enzyme in the catabolism of heme to biliverdin, free iron, and CO. Its expression has been correlated with protection against oxidative stress and ischemia. HO-1 activity is increased in whole animal tissues and in cultured cells following treatment with heme, metals, and inflammatory cytokines, as well as under hypoxic and oxidative conditions. HO-1 is also induced by various stimuli, including heat shock, hyperoxia, and oxidative stress, and represents a powerful endogenous protective mechanism against free radicals in a variety of pathological conditions, including inflammation, ischemic stroke, and many neurodegenerative diseases.7
Hemin is an iron containing metalloporphyrin. Chemically, hemin is represented as chloro (7,12-diethenyl-3,8,13,17-tetramethyl-21H, 23H-porphine 2,18-dipropanoato (2-) -N21, N22, N23, N24) iron. Hemin is substrate for the HO-1 enzyme. HO enzyme is up-regulated by its substrate hemin.8 SnPP is tin-protophophyrin IX, a potent competitive inhibitor of HO, regulates HO by a dual control mechanism that is potently inhibiting the HO enzyme activity while enhancing the synthesis of HO enzyme protein.9
In this study, we examined the expression of HO-1 in lesions of EAU to evaluate the involvement of HO-1 in EAU, and demonstrated that hemin, an inducer of HO-1, ameliorates EAU clinically. To confirm these clinical effects, histological, western blotting, and immunohistochemical analyses of HO-1 were performed.
Materials and Methods
Animals and reagents: A total of 69 adult male Lewis rats with body weights between 150 and 200 g were obtained from SLC, Inc. (Hamamatsu, Japan). All experiments were conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research. Interphotoreceptor retinoid-binding protein (IRBP)-derived peptides were synthesized and purified by Peptron Co., Ltd. (Daejeon, Korea), using the peptide sequence of bovine IRBP determined and reported by Borst et al.10 A epitope of peptide 1169-1191 (PTARSVGAAD……GSSWEGVGVVPDV) was purchased from Sigma (USA). The rats were divided into four groups: 22 served as IRBP-treated controls (IRBP-control group); 22 were treated with the HO-1 inducer, hemin (hemin-treated group); and 22 were treated with the HO-1 inhibitor, tin protoporphyrin IX (SnPP, SnPP-treated group). Other three rats were used normal control for Western blotting and ELISA assay of HO-1.
Immunization (EAU induction with IRBP) and hemin or SnPP treatments: The rats were immunized by subcutaneous injections of 150 g of IRBP in 100 L of emulsion with complete Freund's adjuvant (1:1.5, v/v) that had been supplemented with Mycobacterium tuberculosis to a final concentration of 2.5 mg/mL, without pertussis toxin. Animals were given either 40 mol/Kg hemin (Frontier Scientific Inc., USA), or 40 mol/Kg SnPP (Frontier Scientific Inc.) by intraperitoneal injection once daily for 5-20 days after immunization. IRBP control rats received injections of the same volume of saline on the same schedule. The volume injected into each rat was 0.5 mL.
Clinical assessment of EAU: The incidence and severity of EAU were examined with a biomicroscope on day 3 of the pre-clinical stage of the disease, and thereafter to assess disease development. The clinical signs of inflammation were scored on grades of 0 to 4. Grade 0 indicated no disease; grade 1 indicated enlargement of the iris vessel; grade 2 indicated cellular infiltrates in the anterior chamber; grade 3 indicated fibrin deposition around the pupil and little hypopyon; and grade 4 indicated fibrin plugging of the pupil and development of severe inflammatory hypopyon.11
ELISA assay of HO-1: Three rats of each group were killed in the early stage of the disease (6th day), during peak disease activity (12th day), in the late stage of EAU (18th day) and three rats as normal controls. One eye was for ELISA and the other eye is for Western blot of HO-1. Cornea, aqueous humor and lens were removed and a remnant in the scleral shell was emulsified. The emulsion was used to assay ELISA kit following the protocol. Briefly, The specimen was homogenized, extraction buffer and centrifuged, and the supernatant was used for the assay. One of the rat monoclonal anti-heme oxygenase-1 was immobilized onto the microtiter plate and blocked against non-specific binding. Samples and standards were incubated in the microtiter plate. The second step is to wash the plate and add HO-1 labeled with peroxidase (POD). During this incubation, rat HO-1 is bound to anti-HO-1 on one side and tagged on the other by POD-anti-HO-1. The reaction between POD and substrate results in color development with intensities proportional to the amount of rat HO-1 present in samples and standards. The amount of rat HO-1 was measured the absorbance using an EIA plate reader.
Western blotting analysis: At the same time as the ELISA specimens were prepared, the other eyes of the rats were prepared for western blotting analysis. The specimens were homogenized in lysis buffer containing protease inhibitors,12 and the lysate collected for western blotting analysis. Protein levels were determined by immunoblotting with antibodies directed against mouse HO-1. Briefly, 80 µg of lysate supernatant was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to nitrocellulose membrane (Amersham, Piscataway, NJ) using a semidry transfer apparatus (Bio-Rad, Hercules, CA). The membranes were incubated at 4℃ overnight with 5% milk in 10 mM Tris-HCl (pH 7.4), 150 mM NaCl, and 0.05% Tween 20 (TBST buffer). After the membranes were washed with TBST, they were incubated with constant shaking for 1 h at room temperature with a 1:1,000 dilution of anti-HO-1 antibody. Mouse anti-HO-1 antibodies were obtained from StressGen Biotechnologies Corp. (Victoria, BC, Canada). The membranes were then washed and probed with horseradish-peroxidase-conjugated donkey anti-mouse IgG (Amersham) at a dilution of 1:2,000. Chemiluminescent detection was performed with the Amersham ECL detection kit according to the manufacturer's instructions.
Histologic and immunohistochemistry: One rat of each groups at the stage of peak disease were prepared for histology and immunohistochemistry. Formalin-fixed and paraffin-embedded tissue specimens were cut to a thickness of 4 µm. HO-1 was detected in the tissue specimens described above with a monoclonal antibody (StressGen Biotechnologies Corp.) directed against HO-1. Briefly, paraformaldehyde-fixed tissue sections were thawed and incubated in excess enzyme substrate (3% H2O2 for peroxidase) for 10 min at room temperature. The primary antibody was applied at a dilution of 1:200 overnight at 4℃ and incubated with a streptavidin-conjugated secondary antibody. HO-1 was counter-stained with Gill's hematoxylin for 3 seconds.
Statistical analysis: Clinical uveitis grading was evaluated by Chi-square test and a value of p<0.05 was considered statistically significant.
Results
Effects on Clinical Symptoms of EAU
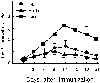
The clinical symptoms of EAU were evaluated in the three groups on alternate three days over the entire 21 days. As shown in Fig. 1, EAU was apparent on day 6 in the SnPP-treated group, reached a state of peak uveitis on day 12, and then eventually diminished until day 21. The hemin-treated group had a lower grade of inflammation than the IRBP-control group; the SnPP-treated group had a higher grade than the IRBP-control group. Each group has 22 rats until 6th day, 19 rats from 9th to 18th day and 15 rats at 21st day. Clinical signs of uveitis were statistically significantly different between SnPP- treated group and IRBP-control group from 6days after immunization. And there were significant difference between Hemin-treated group and IRBP-control group only from 12 days to 18 days after immunization (Fig. 1).
Western blotting of HO-1
HO-1 was strongly expressed in the hemin-treated group at 12th peak day and 18th day. However, HO-1 was less expressed in the SnPP-treated group than in the IRBP-treated group. And IRBP-treated control group was more expressed than normal control due to inflammation response. SnPP treated group has been similarly expressed to normal control (Fig. 2).
ELISA assay of HO-1
This test was performed three rats on each groups on 6th, 12th, and 18th days after IRBP immunization. In a Hemin-treated rat, HO-1 was markedly high compared to an IRBP-control rat, whereas in an SnPP-treated rat, HO-1 was below to the level of the IRBP-control rats (Fig. 3).
Immunohistochemical Detection of HO-1
In the IRBP-control rat, HO-1 was occasionally expressed in the ganglion cell layer and inner nuclear layer. In the hemin-treated rat, HO-1 was expressed more strongly in the ganglion cell layer and the inner nuclear layer than in the IRBP-control rat. There was scanty expression of HO-1 in the SnPP-treated ratIn the IRBP-treated control group. In the normal control, there was no HO-1 expression (Fig. 4).
Discussion
The uveitis is an intractable disease in the field of ophthalmology. Steroids, anti-inflammatory agents or immunosuppressive agents have been used to reduce the uveitis inflammation, even though they have many complications.13 For a new therapeutic modality, exact mechanism of uveitis and animal model of uveitis might be established.
Our results clearly show that HO-1 is expressed in EAU. As in other diseases, HO-1 plays a protective role in EAU. Hemin, an inducer of HO-1, ameliorated the clinical signs of EAU. In contrast, SnPP-treated rats have more severe clinical signs of inflammation especially at the peak period of the disease. These results demonstrate that HO-1 plays an important protective role in EAU.
Over the past decade, a kind of small heat shock protein, HO-1, has been implicated as a cytoprotective enzyme against oxidative injuries.14 HO-1 is well known as the 32-kDa heat shock protein.15 It is now considered a powerful cellular defense mechanism against oxidative stress,16 and plays a protective role in many stress-mediated diseases. For example, a recent experiment demonstrated that HO-1 induction attenuates experimental Parkinson's disease.17 The antioxidant effects of HO-1 are said to be multifactorial and dependent on the diverse dimensions of HO activity. HO-1 catalyzes the rate-limiting step in heme catabolism, generating free iron, CO, and bilirubin.18 Bilirubin is regarded today as a potent antioxidant substance in vitro and as a very effective physiological antioxidant in vivo. Bilirubin scavenges superoxide and peroxyl radicals,19 and possesses antimutagenic and anticomplement properties.20 CO, another heme metabolite that is considered a gaseous messenger, also has anti-inflammatory effects involving the mitogen-activated protein (MAP) kinase pathway.21 CO also inhibits the release of some cytotoxic cytokines.22 HO-1-mediated heme degradation also appears to act in the negative feedback regulation of the production of iNOS-derived NO, and ameliorates the cytotoxicity caused by the free radical NO and its reactive derivative peroxynitrate (ONOO-).23 All these properties contribute to the potent antioxidant effects of HO-1. A variety of stimuli, including oxidative stress, ischemia reperfusion, endotoxin, and cytokines, has been shown to induce HO-1 expression in various tissues. HO-1 catalyzes the rate-limiting step in the degradation of heme to yield equimolar amounts of biliverdin, carbon monoxide (CO), and iron.15 Recently, it has been reported that HO-1 is increased in iris-ciliary body (ICB) of EIU rat.6
A central question is why HO-1 is important in EAU. Oxidative stress resulting from the toxic effects of reactive oxygen species plays a remarkably common role in the mechanisms of a wide variety of diseases.24 There is evidence that oxidative stress is important in the pathogenesis of experimental autoimmune encephalitis (EAE) and multiple sclerosis.25 HO-1 is also considered a factor in the formation and resolution of inflammatory autoimmune lesions of the nervous system.26 In fact, the majority of reports concerning HO and autoimmunity have focused on HO-1.
This study shows that HO-1 is induced in neural retina especially ganglion cell and inner nuclear layer when hemin was introduced. Hemin-induced HO-1 plays an protective role in the EAU animal model. And SnPP makes the uveitis inflammation signs aggravate clinically and that was supportive to other experiments. However, further study would be needed to confirm the role of HO-1 on EAU by statistically proved methods. In conclusions, we have shown the expression of HO-1 in EAU and demonstrated its protective role in this disease, especially at the peak of the illness.



 XML Download
XML Download